Chapter 17
Troubles de l’hémostase et angiœdème héréditaire
Veuillez également consulter le Chapitre 5 : Concentrés pour les troubles de l’hémostase et l’angiœdème héréditaire pour obtenir de l’information sur les concentrés de facteurs de coagulation offerts au Canada.
Saignements anormaux
Des saignements anormaux peuvent résulter d’anomalies des plaquettes, des facteurs de coagulation ou des vaisseaux sanguins/tissus conjonctifs. Ce chapitre porte sur la diathèse hémorragique congénitale et acquise, et met l’accent sur l’utilisation de produits sanguins spécialisés, de produits de protéines purifiées dérivées du plasma et d’autres nouveaux agents.
Le recours à un outil d’évaluation des saignements (ou BAT pour bleeding assessment tool), tel que le système de score de la Société internationale sur la thrombose et l’hémostase (ISTH, International Society on Thrombosis and Haemostasis) (disponible dans la bibliographie1,2), est recommandé pour obtenir les antécédents hémorragiques systématiques permettant de définir le phénotype de saignement d’un patient et d’orienter les investigations (consulter également les figures concernant les outils d’évaluation des saignements et leur mode d’emploi dans l’ouvrage de révision illustré de Elbaz et Sholzberg3). Les épreuves courantes de dépistage de saignements anormaux englobent le temps de céphaline activée (voir la figure sur le temps de céphaline activée dans Elbaz et Sholzberg3), le temps de prothrombine/rapport international normalisé (RIN) (voir la figure sur le temps de prothrombine dans Elbaz et Sholzberg3) et la numération plaquettaire. Néanmoins, ces tests ne permettent souvent pas de détecter la maladie de von Willebrand, trouble congénital de la coagulation le plus fréquent ayant une prévalence d’une personne sur 1 0003,4. La thrombocytopénie est le trouble plaquettaire le plus répandu et est généralement acquise. Des anomalies congénitales affectant la qualité des plaquettes peuvent également survenir et s’accompagner d’une thrombocytopénie. Chez les patients présentant une anomalie plaquettaire, le temps d’occlusion constaté au moyen d’un analyseur de la fonction plaquettaire (PFA-100/200) peut être allongé. Les anomalies vasculaires ou du tissu conjonctif peuvent s’accompagner d’une hyperflexibilité des articulations et/ou d’une hyperlaxité cutanée. Le traitement efficace des troubles de l’hémostase requiert un diagnostic exact ainsi que des épreuves de l’hémostase pointues qui peuvent comprendre le dosage des facteurs de coagulation et des inhibiteurs ainsi que l’étude de la fonction plaquettaire et une analyse des mutations. L’approche algorithmique du diagnostic3,5 déborde le cadre du présent document.
En dehors de la maladie de von Willebrand, les troubles de la coagulation héréditaires ou congénitaux sont relativement rares. Leur traitement peut s’avérer complexe et il vaut mieux privilégier une coordination avec les programmes régionaux de prise en charge de l’hémophilie et des troubles de la coagulation et les hématologues possédant une expertise en la matière. Au Canada, presque toutes les personnes atteintes d’hémophilie sont inscrites à l’un des 25 programmes de prise en charge de l’hémophilie et des troubles de la coagulation en place un peu partout au pays (les coordonnées des centres de traitement se trouvent sur le site Web de la Société canadienne de l’hémophilie6). De plus, les patients inscrits et suivis dans ces centres ont reçu la carte Facteur d’abord (carte Facteur d’abord / FactorFirst wallet card pour l’hémophilie et la maladie de von Willebrand) ou la carte Traitement d’abord (carte Traitement d’abord / TreatmentFirst wallet card pour les troubles héréditaires rares de la coagulation) qui comporte des renseignements importants sur le diagnostic de leur maladie, les soins d’urgence à leur prodiguer en cas d’hémorragie « grave » ou « modérée/légère », de même que les coordonnées du centre où ils sont traités. On recommande aux patients d’avoir cette carte sur eux en tout temps. Après l’administration de la dose d’urgence initiale, les patients doivent en informer les cliniques de prise en charge de l’hémophilie et des troubles de la coagulation (ou l’hématologue de garde si la clinique est fermée) afin d’obtenir des recommandations concernant les soins continus. Il convient également de consulter ces établissements pour connaître les recommandations en matière de couverture hémostatique pour tout acte chirurgical ou effractif.
1. Troubles de la coagulation congénitaux
Bien que le traitement de ces troubles repose essentiellement sur l’augmentation des taux de facteurs de coagulation à l’aide de concentrés ou d’agents pharmaceutiques, l’emploi adéquat d’agents auxiliaires, dont les antifibrinolytiques comme l’acide tranexamique (voir les figures sur le mécanisme d’action et les indications de l’acide tranexamique dans l’ouvrage de révision illustré de Relke et al.5), la colle de fibrine, la thrombine topique, le GELFOAM (éponge en gélatine absorbable) et les hémostatiques polysaccharidiques microporeuses, est souvent efficace dans les cas d’hémorragie légère à des endroits particuliers. Ces agents auxiliaires, utilisés avec des concentrés de facteurs de coagulation en cas d’hémorragie grave, peuvent accélérer le processus d’hémostase et réduire l’administration globale de concentrés de facteurs de coagulation. Des mesures conventionnelles, comme l’application de pression sur la région touchée, le repos, l’application de glace, l’immobilisation, la compression et l’élévation, doivent être prises au besoin. On doit éviter les agents antifibrinolytiques lorsqu’on utilise des concentrés ayant un potentiel thrombogène, tels que les concentrés de complexe prothrombique activé (CCPa, p. ex. FEIBA ou activité de court-circuitage de l’inhibiteur du FVIII) ou les concentrés de complexe prothrombique (CCP), et lorsqu’on traite des patients dont l’hémorragie se situe dans les voies urinaires supérieures et/ou la cavité thoracique. D’une façon générale, les antifibrinolytiques sont sécuritaires lorsqu’ils sont employés avec du FVII activé recombinant (rFVIIa).
a) Hémophilie
L’hémophilie peut être causée par un déficit en FVIII (hémophilie A, hémophilie classique) ou en FIX (hémophilie B, maladie de Christmas), et son incidence est d’environ un cas pour 5 000 à 7 000 nouveau-nés de sexe masculin. L’hémophilie A est la plus fréquente et représente de 80 à 85 % des cas. Le traitement des hémorragies dépend du type d’hémophilie et de sa gravité ainsi que du siège et de l’ampleur de l’hémorragie7. Les patients atteints d’hémophilie A ou B grave ont un taux de facteur de base inférieur à 1 UI/dl (0,01 UI/ml ou 1 %). Ceux dont le taux de facteur est supérieur à 5 UI/dl (0,05 UI/ml ou 5 %) sont atteints d’hémophilie A ou B légère. La forme modérée se situe entre la forme légère et la forme grave.
I. Desmopressine (DDAVP)
En cas d’hémophilie A légère (activité de FVIII de base supérieure à 5 %) et parfois, en cas d’hémophilie A modérée (activité de FVIII de base entre 1 et 5 %), l’utilisation de la desmopressine (0,3 μg/kg de masse corporelle par voie intraveineuse [i.v.] ou sous-cutanée [s.c.]) suffit pour gérer avec succès des hémorragies légères ou des interventions mineures8,9. Dans la mesure du possible, on doit au préalable faire subir des tests au patient pour s’assurer qu’il répond adéquatement à cette substance10. Comme l’administration répétée de doses rapprochées peut entraîner une tachyphylaxie, on doit recourir, en complément, à des concentrés de facteurs de coagulation si le traitement se prolonge. Les patients sous desmopressine peuvent présenter une rétention aqueuse et une hyponatrémie; ces troubles posent un problème particulier chez les enfants de moins de deux ans, les personnes âgées et les patients ayant un système cardiovasculaire à risque. Il est recommandé de restreindre l’apport de liquides (généralement au plus 1 500 ml pendant 24 heures après une dose pour un adulte, et un volume d’entretien en fonction du poids pour un enfant) et de surveiller le taux de sodium durant l’administration de ce traitement.
II. Concentrés de facteurs de coagulation
Les modalités relatives à un traitement de remplacement du FVIII ou du FIX sont énoncées dans le tableau 1. On y décrit la concentration de pointe des facteurs de coagulation recommandée pour différents types d’hémorragie et le traitement d’entretien dans les cas d’hémorragie grave. La figure 1 présente une méthode de calcul de la dose en UI/kg convenant à ces concentrés (applicable également à d’autres concentrés de facteurs de coagulation) avec des taux de récupération in vivo (RIV) connus. En général, l’intervalle entre deux doses peut être identique à la demi-vie (t1/2) du facteur de coagulation. Dans ce cas, la dose d’entretien nécessaire à l’obtention de la concentration maximale d’origine du facteur de coagulation représente la moitié de la dose d’attaque. Il faudrait procéder à des études pharmacocinétiques visant à mesurer le temps de récupération et la demi-vie afin de déterminer la dose et l’intervalle adéquats, puisque ces paramètres pharmacocinétiques varient d’un produit à l’autre et, pour chaque produit, d’un individu à l’autre. Cet aspect revêt une importance particulière chez les enfants, qui peuvent présenter un plus grand volume plasmatique et avoir ainsi besoin de doses plus élevées pour parvenir aux mêmes taux de facteur de coagulation qu’un adulte. Il est maintenant possible de déterminer les paramètres pharmacocinétiques au moyen de deux à quatre prélèvements seulement, en s’appuyant sur la technique d’analyse pharmacocinétique de population offerte par WAPPS-Hemo11.
La perfusion continue après l’administration d’une dose d’attaque lors d’une hémorragie grave ou d’une intervention chirurgicale (voir la figure 1) est avantageuse en ce qu’elle permet d’obtenir un taux de facteur de coagulation in vivo plus constant, dépourvu des pics et des creux résultant de l’administration de bolus intraveineux. La perfusion continue peut se traduire par une utilisation globale moindre de concentrés. Il faut utiliser des pompes à perfusion pouvant administrer de petites quantités, car les concentrés ne doivent pas être dilués au-delà des seuils recommandés par le fabricant. Il faut surveiller attentivement la pompe, le site d’injection et la tubulure pour s’assurer de l’absence d’interruption dans le traitement.
En ce qui a trait au concentré de FIX, on recommande d’administrer environ les dix à vingt premières perfusions aux personnes qui viennent de recevoir un diagnostic d’hémophilie B grave dans un établissement équipé pour la prise en charge des réactions allergiques graves7. En effet, jusqu’à 5 % des personnes atteintes d’hémophilie B grave peuvent développer des inhibiteurs (généralement à un stade précoce du traitement de remplacement du FIX), qui s’accompagnent souvent de graves réactions allergiques, y compris l’anaphylaxie7.
Tableau 1 : Concentration plasmatique de pointe des facteurs de coagulation et durée d’administration recommandées pour le traitement d’hémorragies et la prévention en cas d’interventions chirurgicales (pour des précisions sur les concentrés de facteurs de coagulation offerts, consulter le tableau 1 du Chapitre 5 du présent Guide)
| Type d’hémorragie | Hémophilie A | Hémophilie B | ||
|---|---|---|---|---|
| Concentration de pointe désirée (UI/dl)† | Durée (en jours) | Concentration de pointe désirée (UI/dl)* | Durée (en jours) | |
| Articulation | 40–60 | 1-2 Le traitement peut être prolongé si la réponse est inadéquate. |
40–60 | 1-2 Le traitement peut être prolongé si la réponse est inadéquate. |
|
Douleurs musculaires |
40–60 |
2-3 Le traitement peut être prolongé si la réponse est inadéquate. |
40–60 |
2-3 Le traitement peut être prolongé si la réponse est inadéquate. |
|
Iliopsoas
|
80–100 |
1–2 |
60–80 |
1–2 |
|
SNC ou tête
|
80–100 |
1–7 |
60–80 |
1–7 8–21 |
|
Gorge et cou
|
80–100 50 |
1–7 8–14 |
60–80 30 |
1–7 8–14 |
|
Appareil digestif
|
80–100 50 |
1–7 7–14 |
60–80 30 |
1–7 7–14 |
| Reins | 50 | 3–5 | 40 | 3–5 |
| Lacération profonde | 50 | 5–7 | 40 | 5–7 |
|
Chirurgie (majeure)
|
80–100 |
s.o. |
60–80 |
s.o. |
|
Extraction dentaire‡
|
30–50 |
s.o. |
30–50 | s.o. |
|
D’après les Lignes directrices pour la prise en charge de l’hémophilie de la FMH7 |
||||

Remarque : Il s’agit de calculs de posologie pratiques, effectués à partir des valeurs de récupération et des valeurs moyennes de la demi-vie. La posologie est plus exacte si l’on se fonde sur les paramètres pharmacocinétiques du produit en question pour un patient donné. Une évaluation simplifiée des paramètres pharmacocinétiques de population basée sur un échantillonnage épars est proposée par WAPPS-Hemo7.
*RIV = récupération in vivo (hausse) de l’activité en UI/dl selon la quantité perfusée en fonction de la masse corporelle; se reporter au tableau 1 du chapitre 5 du présent Guide pour connaître la récupération in vivo moyenne de divers produits de FVIII et de FIX.
III. Prévention de saignements7,12,13
a) Prophylaxie : introduction
La prophylaxie est considérée comme la norme en matière de prise en charge des patients atteints d’hémophilie A ou B grave et de ceux présentant des phénotypes de saignements graves sur le plan clinique, quels que soient les taux mesurés de facteurs de coagulation.
- Par rapport au traitement de remplacement épisodique, la prophylaxie empêche la survenue de saignements engageant le pronostic vital, réduit l’invalidité chronique due aux saignements intramusculaires récurrents et aux hémarthroses et améliore la qualité de vie des patients.
- La prophylaxie consiste à administrer de manière régulière et continue un ou plusieurs agents hémostatiques dans le but de prévenir les saignements chez les personnes atteintes d’hémophilie tout en leur permettant de mener des vies actives et d’avoir une qualité de vie comparable aux non-hémophiles.7
- Classiquement, l’objectif de la prophylaxie est de faire en sorte qu’une personne atteinte d’hémophilie grave (taux de FVIII ou FIX < 1 UI/dl) présente un phénotype de saignement se rapprochant de celui d’une personne atteinte d’hémophilie modérée en maintenant des taux de facteurs de coagulation > 1 UI/dl.
- Néanmoins, il est de plus en plus admis et éprouvé que des taux minimaux de facteurs de coagulation entre 1 et 3 UI/dl ne permettent pas d’éviter tous les saignements. En fonction du niveau d’activité physique, du statut articulaire et du phénotype de saignement d’une personne, des taux supérieurs peuvent en effet être nécessaires pour éviter les saignements cliniques et subcliniques ainsi que la progression de maladie articulaire.
- Des études épidémiologiques sur l’hémophilie non grave ont montré que chaque hausse de 1 % des taux de facteurs de coagulation de base est associée à une baisse de la fréquence hémorragique; au-delà de 15 UI/dl, les saignements spontanés sont rares14.
- Par conséquent, l’objectif de la prophylaxie est désormais de limiter autant que possible les saignements de façon à prévenir la survenue (chez les enfants) ou la progression (chez les adultes) de l’arthropathie hémophilique, la plupart des cliniciens visant de préférence des taux minimaux plus élevés (> 3-5 %).
- Il est possible d’atteindre cet objectif en employant une approche personnalisée ou sur mesure de la prophylaxie (voir section suivante). L’utilisation de concentrés de facteurs de coagulation à demi-vie prolongée, mais aussi de concentrés de facteurs de coagulation à demi-vie normale à des doses supérieures ou à une fréquence accrue ainsi que des traitements non axés sur les facteurs de coagulation sont autant de stratégies possibles.
b) Définitions7
Prophylaxie primaire : On amorce un traitement continu régulier chez les patients de moins de trois ans, avant la deuxième hémorragie dans les grosses articulations (coude, genou ou cheville) et en l’absence d’ostéochondrite.
Prophylaxie secondaire : Le traitement continu régulier est amorcé à la suite d’au moins deux hémorragies dans de grosses articulations, mais avant le début d’une ostéochondrite, et généralement à partir de l’âge de trois ans.
Prophylaxie tertiaire : Le traitement continu régulier est amorcé après le début de l’arthropathie documentée. La prophylaxie tertiaire correspond généralement à une prise en charge amorcée à l’âge adulte.
Prophylaxie intermittente : Le traitement est administré pour prévenir les saignements pendant moins de 45 semaines par an. (La prophylaxie est, par exemple, conseillée avant la pratique d’activités et à la suite d’une grave hémorragie afin de prévenir les saignements récurrents, ou après une intervention chirurgicale afin de prévenir la survenue d’un saignement postopératoire, ou après la survenue d’un nouvel épanchement de sang dans une même articulation [articulation cible] pour rompre le cercle vicieux des saignements.)
Traitement de remplacement épisodique (sur demande) : Des concentrés de facteurs de coagulation sont uniquement administrés au moment du saignement.
c) Prophylaxie traditionnelle à base de concentrés de facteurs de coagulation
- Une prophylaxie à doses élevées ou intermédiaires amorcée à un jeune âge est associée à une réduction de plus de 90 % des taux de saignements articulaires et à une réduction significative des maladies articulaires dégénératives.
- La plupart des études menées avec des concentrés de facteurs de coagulation se basent sur des protocoles de prophylaxie fixes/non personnalisés.
- Prophylaxie à doses élevées : 25-40 UI de FVIII/kg tous les deux jours, 40-60 UI de FIX/kg deux fois par semaine
- Prophylaxie à doses intermédiaires : 15-25 UI de FVIII/kg trois jours par semaine, 20-40 UI de FIX/kg deux fois par semaine
- Prophylaxie à faibles doses : 10-15 UI de FVIII/kg deux à trois jours par semaine, 10-15 UI de FIX/kg deux jours par semaine12,15-17
- Avec une prophylaxie basée sur des concentrés de facteurs de coagulation à demi-vie normale administrés à doses fixes, il est difficile d’atteindre des taux minimaux de facteurs de coagulation dépassant 1 %. Étant donné la courte demi-vie de ces produits, il est nécessaire d’effectuer fréquemment des ponctions veineuses pour assurer une prophylaxie adéquate. Seules des perfusions très fréquentes (c’est-à-dire quotidiennes) permettent éventuellement d’atteindre des taux minimaux supérieurs avec des produits à demi-vie normale.
- Les concentrés de facteurs de coagulation à demi-vie prolongée permettent d’atteindre des taux minimaux supérieurs avec moins de perfusions. De ce fait, il n’est pas forcément nécessaire d’installer un dispositif d’accès veineux central pour faciliter la prophylaxie chez les jeunes enfants, et ces produits peuvent aussi améliorer l’observance du traitement chez certains patients.
d) Prophylaxie personnalisée/sur mesure à base de concentrés de facteurs de coagulation
- Les protocoles de prophylaxie personnalisés/sur mesure sont ajustés aux besoins de chaque patient avec comme objectif commun, l’absence de saignements spontanés.
- La prophylaxie adaptée à une personne donnée déprendra du concentré de facteur de coagulation utilisé, du profil pharmacocinétique du produit chez la personne concernée et de considérations cliniques telles que le niveau d’activité physique, le statut articulaire et le phénotype de saignement.
- En se basant sur des outils d’estimation des données pharmacocinétiques, comme ceux offerts par WAPPS-Hemo11, il est possible d’ajuster la dose et la fréquence du traitement prophylactique en fonction des paramètres pharmacocinétiques de l’individu et du concentré de facteur de coagulation en vue d’atteindre la concentration minimale cible. La prophylaxie basée sur les données pharmacocinétiques peut aussi fournir des indications sur le type de concentrés de facteurs de coagulation (p. ex. produits à demi-vie normale ou prolongée) le plus adapté pour atteindre les objectifs cliniques et les taux minimaux pour un individu donné. Par ailleurs, il convient d’adapter la prophylaxie personnalisée en fonction de facteurs cliniques comme le phénotype de saignement et les tendances en matière d’activité physique.
- La dose et la fréquence doivent être ajustées (vers le haut ou le bas) afin de supprimer les saignements cliniques excessifs grâce à une prophylaxie d’intensité minimum.
e) Traitement préventif non axé sur des facteurs de coagulation
Il s’agit de médicaments/produits permettant l’hémostase grâce à un mécanisme différent de celui des produits de remplacement du FVIII/FIX traditionnels.
- Au moment de la publication du présent chapitre, le seul produit non axé sur des facteurs de coagulation autorisé pour le traitement de l’hémophilie A est l’émicizumab (Hemlibra®).
- Il imite l’action du FVIII en liant le FIXa et le FX et en activant le FX en FXa (d’où l’effet mimétique du FVIII) pour améliorer l’hémostase. Au Canada, l’émicizumab est approuvé par Santé Canada pour l’indication suivante : prophylaxie de routine visant à prévenir les saignements et à réduire la fréquence des épisodes hémorragiques en cas d’hémophilie A (déficit congénital en FVIII) présentant ou non des inhibiteurs du FVIII. Il est désormais pris en charge et offert par la Société canadienne du sang et Héma-Québec pour la prophylaxie de routine chez les patients atteints d’hémophilie A congénitale présentant des inhibiteurs du FVIII ainsi que chez les patients atteints d’hémophilie A grave sans inhibiteurs.
- Contrairement à la prophylaxie classique à base de concentrés de facteurs de coagulation, l’émicizumab ne remplace pas le facteur de coagulation manquant et peut être administré par voie sous-cutanée à une fréquence moindre (p. ex. toutes les semaines ou toutes les deux ou quatre semaines).
- De plus, l’émicizumab ne suit pas les courbes maximales et minimales des paramètres pharmacocinétiques des traitements par des concentrés de facteurs de coagulation (voir le chapitre 5 du présent Guide).
- Des études cliniques ont démontré l’innocuité et l’efficacité de l’émicuzimab (Hemlibra®) pour la prophylaxie de l’hémophilie et la protection contre les saignements (taux de saignement annualisé [TSA] de 1,3-1,5)18,19.
- L’administration sous-cutanée et les perfusions peu fréquentes ont l’avantage d’offrir une prophylaxie moins contraignante, ce qui devrait faciliter l’instauration du traitement à un âge plus jeune.
- Pour le traitement de métrorragies survenant sous prophylaxie par l’émicizumab, il convient de réaliser des perfusions de FVIII aux doses habituellement administrées pour atteindre l’hémostase7,20.
- MISE EN GARDE concernant la surveillance de la coagulation dans le cadre d’une prophylaxie par l’émicizumab :
- Le temps de céphaline activée sera faussement raccourci et ne doit pas être utilisé pour le suivi, sauf dans les rares cas où l’on soupçonne le développement d’anticorps anti-émicizumab (le temps de céphaline activée sera prolongé de manière inappropriée en l’absence d’effets hémostatiques de l’émicizumab)7,20. Étant donné que l’émicizumab met du temps à disparaître, son influence sur le raccourcissement du temps de céphaline activée persistera jusqu’à six mois après l’arrêt du traitement.
- Le dosage du FVIII et des inhibiteurs du FVIII doit être effectué à l’aide d’une méthode chromogénique utilisant des réactifs bovins7,20. L’interférence de l’émicizumab avec les tests basés sur le temps de céphaline activée persistera jusqu’à six mois après l’arrêt du traitement.
- D’autres traitements non axés sur le remplacement de facteurs de coagulation sont en cours de développement et incluent des agents qui inhibent/suppriment les anticoagulants endogènes naturels (antithrombine, inhibiteur de la voie du facteur tissulaire [TFPI pour tissue factor pathway inhibitor] et protéine C activée, se reporter à la section sur les nouveaux traitements et innovations dans la prise en charge de l’hémophilie).
f) Programmes d’exercices pour la prévention de saignements
Les patients devraient suivre un programme d’exercices tenant compte de l’état de leur appareil locomoteur afin d’améliorer leur tonus musculaire et leur équilibre, de maintenir une bonne condition physique pour avoir une bonne santé générale et d’éviter les blessures et les saignements. La pratique d’exercices de mise en charge peut également améliorer la densité osseuse et ainsi prévenir l’ostéopénie ou l’ostéoporose, des affections prédisposant les patients aux fractures de fragilisation.
b) Hémophilie avec inhibiteurs et inhibiteurs du FVIII acquis21,22
Des inhibiteurs (anticorps) de la protéine du facteur de coagulation déficitaire apparaissent chez 20 à 30 % des personnes atteintes d’hémophilie A grave et jusque chez 5 % des personnes atteintes d’hémophilie B7. Les patients atteints d’hémophilie A légère/modérée présentent également un risque à vie de développer des inhibiteurs en cas d’exposition croissante au FVIII exogène, l’incidence cumulative étant de 5,3 % après 28 jours d’exposition et passant à 13,3 % après 100 jours d’exposition23. La présence d’inhibiteurs entraîne un risque de saignements accru et rend difficile un traitement à l’aide de concentrés de facteurs de coagulation. Chez ces patients, le traitement de l’hémorragie doit s’effectuer de concert avec un établissement ayant de l’expérience dans la prise en charge des hémophiles présentant de tels inhibiteurs. Toutes les hémorragies graves doivent être traitées dans ces établissements.
I. Hémophilie A avec inhibiteurs du FVIII
a) Traitement de saignements aigus
- Chez les patients dont le titre d’inhibiteurs est faible au moment du traitement (˂ 5 UB), on peut administrer une dose de concentré de facteur de coagulation humain suffisamment élevée pour neutraliser les inhibiteurs, mais déployant une activité excédentaire permettant d’arrêter l’hémorragie. On peut entreprendre le traitement à l’aide de doses de 100 UI/kg en surveillant la réponse clinique et le taux du facteur de coagulation afin de modifier la dose au besoin.
- Les patients ayant un taux d’inhibiteurs compris entre 5 et 10 UB sont peu susceptibles de répondre à des concentrés de FVIII. Parmi les autres agents « de dérivation » parmi lesquels choisir figurent le rFVIIa (Niastase; environ de 90 à 120 µg/kg toutes les 2 à 3 heures) et le FEIBA® (de 50 à 100 U/kg toutes les 8 à 12 heures, sans dépasser 200 U/kg par 24 h).
- Les antifibrinolytiques peuvent être administrés comme traitement complémentaire avec le FVIII d’origine humaine et avec le rFVIIa, mais ils doivent être évités lors de l’administration du FEIBA®. On doit prévoir un délai de 3 à 6 heures lorsqu’on passe du rFVIIa au FEIBA®, et de 6 à 12 heures lors du passage du FEIBA® au rFVIIa, afin de réduire le potentiel thrombogène de leur association. On a fait état d’une étude de série de cas où ces deux agents ont été utilisés ensemble avec succès (chacun administré à dose réduite)24.
- Chez les patients réfractaires (chez qui on ne parvient pas à contenir une hémorragie grave), il peut être nécessaire d’effectuer une plasmaphérèse ou d’avoir recours à une immuno-adsorption sur colonne pour enlever les IgG plasmatiques (uniquement dans certains centres de traitement) afin d’abaisser rapidement le titre des inhibiteurs et de favoriser une utilisation efficace des concentrés contenant du FVIII. Le FVIII porcin recombinant constitue une autre option en cas d’hémorragie potentiellement mortelle (p. ex. SNC) ou si une opération est requise25,26. Cependant, au moment de la rédaction du présent chapitre, le FVIII recombinant d’origine porcine n’est autorisé qu’en cas d’hémophilie acquise et pas encore en cas d’hémophilie congénitale avec inhibiteurs.
b) Prévention (prophylaxie) de saignements
- La prévention (prophylaxie) de saignements chez des patients atteints d’hémophilie A avec inhibiteurs peut être réalisée à l’aide de l’émicizumab (Hemibra®) offert par la Société canadienne du sang et Héma-Québec. L’émicizumab est un anticorps bispécifique des facteurs de coagulation humains IX/IXa et X/Xa qui imite l’activité du FVIII et peut être administré par voie sous-cutanée à intervalles de une à quatre semaines19.
- Des études cliniques ont démontré la supériorité de l’émicizumab par rapport aux agents de dérivation précédemment utilisés (administration de rFVIIa tous les jours ou de FEIBA® tous les deux jours) pour la prévention de saignements chez des patients avec inhibiteurs du FVIII27.
- Bien que la prophylaxie par l’émicizumab soit efficace en cas d’hémophilie A avec inhibiteurs, des métrorragies peuvent toujours survenir et nécessiter un traitement par des concentrés de facteurs de coagulation traditionnels. La prudence est de mise lorsque l’on a recourt à des concentrés de facteurs de coagulation (concentré de complexe prothrombique activé [CCPa]/FEIBA® notamment) pour traiter des saignements survenant chez des patients sous émicizumab, dans la mesure où des cas de microangiopathie thrombotique et de thrombose ont été signalés dans les études cliniques. Si un concentré de CCPa/FEIBA® doit être administré dans le cadre d’une prophylaxie par l’émicizumab, la posologie ne doit pas dépasser 50 UI/kg/dose et 100 UI/kg/dose totale sur 24 heures20. Le rFVIIa apparaît sécuritaire dans le cadre d’une administration concomitante à l’émicizumab.
c) Éradication des inhibiteurs du FVIII
- On peut tenter d’éradiquer les inhibiteurs à l’aide d’un traitement par induction de tolérance immunitaire par perfusions régulières de FVIII tous les jours ou tous les deux jours. L’induction de tolérance immunitaire est un traitement particulièrement contraignant du fait de l’intensité et de la fréquence des perfusions de FVIII et nécessite souvent l’installation d’un dispositif d’accès veineux central chez les jeunes enfants.
- Avec l’émicizumab désormais offert et constituant une option plus efficace que les agents de dérivation pour prévenir les saignements en cas d’hémophilie A avec inhibiteurs, le besoin d’instaurer un traitement par induction de tolérance immunitaire fait aujourd’hui l’objet de débats. Des études cliniques [NCT04023019]28 sont en cours pour déterminer si l’association d’une induction de tolérance immunitaire et de l’émicizumab peut offrir de meilleurs résultats cliniques que l’induction de tolérance immunitaire seule ou la prophylaxie par l’émicizumab seule.
II. Hémophilie B avec inhibiteurs du FIX
Le principe thérapeutique est semblable à celui appliqué pour l’hémophilie A avec inhibiteurs.
- Il faut toutefois savoir qu’environ 50 % des hémophiles de type B avec inhibiteurs peuvent manifester des réactions allergiques graves (dont l’anaphylaxie) aux concentrés contenant du FIX et au FEIBA®. Chez ces patients, on peut utiliser du rFVIIa.
- Le syndrome néphrotique constitue une complication possible/connue chez les patients allergiques présentant des inhibiteurs qui suivent un traitement par induction de tolérance immunitaire par perfusions répétées de concentrés de FIX.
- L’émicizumab n’est PAS efficace chez les patients atteints d’hémophilie B avec inhibiteurs.
III. Inhibiteurs du FVIII acquis
Les inhibiteurs du FVIII acquis, qui sont associés à l’« hémophilie A acquise », constituent un trouble de la coagulation acquis rare (une personne sur cinq millions), mais potentiellement mortel, causé par le développement d’auto-anticorps neutralisants (inhibiteurs) dirigés contre le FVIII29.
- Des saignements cutanéo-muqueux (plus que musculosquelettiques), s’accompagnant notamment d’hématomes cutanés importants, sont souvent les symptômes initiaux d’une hémophilie acquise.
- Cependant, les saignements sont souvent graves et peuvent être fatals.
- Des études d’hémostases montrent un allongement isolé du temps de céphaline activée (du fait d’une baisse ou d’une carence en FVIII), sans correction dans une étude de mélange 1:1 avec « incubation » (37 oC pendant deux heures) et avec confirmation par un dosage positif d’inhibiteurs du FVIII (méthode Bethesda)3.
- La population de patients affectés inclut des patients présentant des maladies auto-immunes sous-jacentes ou des tumeurs malignes, des personnes âgées, des femmes après un accouchement ou des patients sans cause spécifique sous-jacente (environ 50 %)30.
- La prise en charge comprend le traitement concomitant des événements hémorragiques, le traitement d’éradication des inhibiteurs et le traitement des comorbidités (dans la mesure du possible).
a) Traitement de saignements aigus
- Les saignements légers peuvent souvent être contenus avec succès à l’aide de la desmopressine (de 0,3 à 20 µg/kg par voie intraveineuse ou sous-cutanée) et/ou d’autres mesures conservatrices.
- Les hémorragies graves nécessitent l’administration de rFVIIa (environ 90 µg/kg toutes les 2 à 3 heures) ou de FEIBA® (de 50 à 100 U/kg toutes les 8 à 12 heures, pour un maximum de 200 U/kg par jour), sous une surveillance clinique étroite.
- Du FVIII recombinant d’origine porcine (rFVIIIp) peut être utilisé pour le traitement des saignements potentiellement mortels ou menaçant l’intégrité physique des patients et pendant les interventions chirurgicales. La posologie initiale dépend de la réactivité croisée des inhibiteurs du patient au FVIII porcin31,32. Soulignons que l’utilisation de cet agent peut être surveillée grâce aux niveaux d’activité du FVIII31,32. Ce produit doit être utilisé uniquement dans un centre ayant l’expérience de la prise en charge des patients présentant des inhibiteurs.
b) Traitement d’éradication des inhibiteurs
- Il est possible d’instaurer un traitement immunosuppresseur (prednisone à raison de 1 mg/kg par jour seule ou avec d’autres agents tels que le cyclophosphamide à raison de 1-1,5 mg/kg par jour ou rituximab à raison de 375 mg/m2 toutes les semaines pendant quatre semaines) pour éradiquer les inhibiteurs.
- Un traitement prophylactique contre pneumocystis jroveci doit être offert aux patients sous immunosuppresseurs.
c) Traitement des comorbidités
Il convient d’envisager et de gérer la détection et la prise en charge des comorbidités associées à la formation d’inhibiteurs du FVIII.
c) Nouveaux traitements et innovations dans la prise en charge de l’hémophilie
Depuis de nombreuses années, le traitement et la prévention des saignements chez les patients hémophiles sont limités par la courte demi-vie des produits de facteurs de coagulation « endogènes » (aujourd’hui appelés « produits à demi-vie normale ») et la nécessité de perfusions intraveineuses fréquentes. Il existe maintenant des facteurs de coagulation à demi-vie prolongée ainsi que l’émicizumab sur le marché et de nombreux traitements autres que des facteurs de coagulation sont en cours de développement clinique. Les technologies relatives à la demi-vie prolongée des facteurs de coagulation, aux voies d’administration sous-cutanée et à l’atténuation du risque de développer des inhibiteurs ont également accéléré l’exploration de nouveaux traitements.
I. Concentrés de coagulation à demi-vie prolongée33
Des produits à demi-vie prolongée ont été créés à l’aide de la technologie de la fusion, afin de lier une fraction (fragment Fc ou albumine) au facteur de coagulation recombinant endogène, ou par l’ajout de la pégylation à la protéine. Ces deux stratégies prolongent la période pendant laquelle le facteur de coagulation demeure en circulation. Ce type de technologie multiplie par environ 1,5 la demi-vie du FVIII, et par 2,5 à 4,8 celle du FIX34. La prolongation de la demi-vie est propre au produit et au patient, et il est possible d’optimiser la détermination de la dose ou de l’intervalle posologique grâce aux paramètres pharmacocinétiques individuels. Entre autres avantages potentiels, les produits à demi-vie prolongée peuvent permettre une posologie moins fréquente, des taux minimaux plus élevés et un meilleur respect du traitement préventif.
Une prolongation accrue de la demi-vie du FVIII, y compris la fusion avec les polymères protéiques XTEN (rFVIIIFc-VWF-XTEN [BIVV001, Sanofi]) et la rD’D3 VWF-FP (protéine de fusion [albumine] recombinante D’D3 du FvW, CSL626, CSL-Behring) administrés de manière concomitante avec du FVIII exogène, s’avère efficace dans les études cliniques précoces et les études précliniques35,36. Le rFVIIIFc-VWF-XTEN (BIVV001, Sanofi) a achevé des études cliniques de phase I/IIa avec une seule injection intraveineuse et présente une demi-vie moyenne trois à quatre fois plus longue que celle du rFVIII à demi-vie normale37. Après une seule injection, le taux de FVIII était revenu à la normale (≥ 51 UI/dl) pendant 4 jours et se situait à 17 UI/dl au jour 7, suggérant la possibilité d’intervalles hebdomadaires entre les traitements. L’étude de phase III sur le BIVV001 est en cours (NCT04161495).
II. Exploration de la voie d’administration sous-cutanée pour les concentrés de facteurs de coagulation
a) Produits de FIX sous-cutanés
- Le BIVV002 (rFIXFc-XTEN, Sanofi), qui ajoute des polymères XTEN à une molécule du variant Padua du fragment Fc du FIX recombinant (F9R338L), fait l’objet d’études précliniques.
- Le dalcinonacog alfa (DalcA), protéine de FIX modifiée par trois substitutions d’acides aminés en vue d’augmenter l’activité catalytique afin de permettre l’administration sous-cutanée du traitement prophylactique, fait l’objet d’une étude de phase I/II (NCT03186677, NCT03995784)
b) Produits de FVIII sous-cutanés
Le N8-GP s.c. (turoctocog alfa [INN] pegol-sc, Novo Nordisk) a achevé une étude de phase I, mais son développement a été abandonné du fait de la survenue d’anticorps anti-N8-GP.
III. Traitements non axés sur les facteurs de coagulation / traitements de rééquilibrage
L’émicizumab, anticorps bispécifique anti-FIX/FIXa et anti-FX/FXa mentionné précédemment, est le premier traitement non axé sur un facteur de coagulation utilisé en contexte clinique pour la prophylaxie chez des personnes atteintes d’hémophilie A présentant ou non des inhibiteurs du FVIII. Un anticorps bispécifique mimétique de FVIIIa de nouvelle génération présentant une activité apparemment renforcée38 (Mim8, Novo Nordisk) fait actuellement l’objet d’études cliniques de phase III (NCT05306418, NNC0365-3769).
La manipulation des voies pour rééquilibrer l’hémostase ou imiter l’activité du FVIII connaît un développement rapide dans la recherche sur l’hémophilie.
Ces technologies en cours de développement présentent plusieurs avantages : voie d’administration sous-cutanée, hémostase efficace en présence ou en l’absence d’un inhibiteur du facteur de coagulation, et réduction du risque de développement d’inhibiteurs du facteur de coagulation (quoique des anticorps anti‑médicaments soient encore possibles).
- Le fitusiran (ALN-AT3, Sanofi) est un petit ARN interférent (petit ARNi) qui interfère avec la synthèse d’antithrombine. Il diminue donc le taux d’antithrombine endogène, ce qui améliore la production de thrombine et la formation de caillots de fibrine sans avoir à remplacer le FVIII ou le FIX39. Après avoir été temporairement arrêtés du fait d’un cas fatal de thrombose veineuse des sinus cérébraux, des études de phase II ont repris à la suite du déploiement d’une stratégie d’atténuation des risques offrant une orientation sur la posologie et l’utilisation d’agents de dérivation en cas de saignement aigu. Le fitusiran, traitement prophylactique mensuel administré par voie sous-cutanée, a permis de maintenir une baisse de l’antithrombine et d’améliorer la production de thrombine avec un taux de saignement annualisé médian de 1,5 chez les patients de l’étude (NCT02554773)40. Des études de phase III sur le fitusiran administré tous les mois par voie sous-cutanée sont en cours (NCT03417102, NCT03417245, NCT03549871).
- L’inhibiteur de la voie du facteur tissulaire (ou TFPI pour tissue factor pathway inhibitor) joue un rôle inhibiteur important dans la coagulation en régulant l’initiation de la production de thrombine grâce au blocage du complexe facteur tissulaire-FVII et de la prothrombinase. Le blocage du TFPI à l’aide d’anticorps ou d’aptamères est également à l’étude.
- Le concizumab (NN7415, Novo Nordisk) est un anticorps monoclonal IgG4 humanisé dirigé contre le domaine K2 du TFPI. Il a démontré une hausse de la production de pointe de la thrombine41 et a amélioré le taux de saignement annualisé en cas d’hémophilie A, ainsi qu’en cas d’hémophilie A et B avec inhibiteurs dans le cadre d’une administration quotidienne sous-cutanée42. Des études de phase III (NCT04083781, NCT04082429) sont en cours de recrutement.
- Le marstacimab (PF-06741086, Pfizer) est un anticorps monoclonal IgG1 humain ciblant le domaine K2 du TFPI. Une étude multidose de phase Ib/II a montré une baisse du taux de saignement annualisé grâce à des injections hebdomadaires sous-cutanées43. Une étude de phase III avec permutation (NCT03938792) sur le marstacimab administré une fois par semaine par voie sous-cutanée en cas d’hémophilie A ou B grave avec ou sans inhibiteur est en cours de recrutement.
- La protéine C activée est un anticoagulant naturel qui agit en dégradant le FV et le FVIII activés. L’inhibition de la protéine C activée fait l’objet d’examen en tant que modalité de traitement en cas d’hémophilie.
- Le HAPC1573 (mAb, Bayer) est un anticorps monoclonal ciblant la protéine C activée afin d’interférer avec l’inactivation de FVa et FVIIIa. Son administration entraîne le raccourcissement du temps de céphaline activée et l’augmentation de la production de thrombine dans le plasma avec déficit en FVIII et permet de restaurer l’hémostase dans des études précliniques chez les primates44. Les études sur l’être humain n’ont pas encore commencé.
- Le SerpinPC (ApcinteXLtd) est un inhibiteur de la sérine protéase qui a été modifié pour inactiver en particulier la protéine C activée et influer uniquement sur l’activité spécifique de la protéine C activée en matière de coagulation45. Une étude de phase I/II (NCT04073498) est en cours chez des patients atteints d’hémophilie A ou B grave présentant ou non des inhibiteurs.
d) Thérapie génique
La thérapie génique pour l’hémophilie A et B a rapidement progressé. Dans la plupart des cas, on utilise des virus adéno-associés (VAA) comme vecteurs pour apporter le transgène du facteur de coagulation manquant aux hépatocytes. Les technologies de thérapie génique cherchent à pallier différentes limitations, y compris en termes d’efficacité du transfert par le vecteur viral, d’expression à long terme du transgène permettant d’atteindre des taux cliniquement significatifs (fourchette faible/modérée pour le FVIII ou FIX), de perte d’activité liée à la toxicité hépatique après la perfusion du vecteur et d’anticorps neutralisants ciblant les vecteurs à VAA.
I. Thérapie génique en cas d’hémophilie B
- Les données à long terme connues sur l’essai de thérapie génique utilisant l’adénovirus de sérotype 8 et le gène FIX manquant montrent des taux de FIX soutenus (moyenne de 5,1 UI/dl) pendant une période pouvant atteindre sept ans46.
- La thérapie génique faisant appel au vecteur du VAA recombinant (Spark-9001)-FIX Padua (variant FIX Arg338Leu à gain de fonction) a donné lieu à un taux de FIX soutenu moyen de 33,7 UI/dl pendant une période allant de 28 à 78 semaines47. L’étude de phase III utilisant ce vecteur (PF-06838435/fidanacogène élaparvovec/ RAAV-SPARK100-hFIX-Padua, Pfizer) est actuellement en phase de recrutement dans le but d’évaluer de manière plus approfondie l’efficacité et l’innocuité.
- Une étude de phase I/IIb de conception similaire, mais utilisant le transgène du variant Padua du FIX à codon optimisé (AAV5-hFIXco-Padua/AMT-061, UniQure) fait état d’une activité moyenne de FIX à 38 UI/dl après douze semaines. Un rapport intermédiaire sur l’étude de phase III (NCT03569891) incluant des patients avec anticorps neutralisants anti-AAV5 préexistants a montré une activité moyenne de FIX à 37,2 UI/dl après 26 semaines et aucun effet des anticorps neutralisants anti-AAV5 sur le taux de FIX.
- Une étude de confirmation de dose de phase I/II porte sur le FLT-180a (Freeline) avec un vecteur à VAA incapable de se répliquer (NCT03369444) et une autre étude (NCT01687608) utilise le variant Padua du FIX avec un VAA de sérotype 8 optimisé auto-complémentaire (AskBio009, Shire/Takeda).
II. Thérapie génique en cas d’hémophilie A
- L’étude de phase III sur le FVIII avec un VAA de sérotype 5 (AAV5-hFVIII-SQ, également connu sous le nom de valoctocogène roxaparvovec, Biomarin) a montré des taux de FVIII soutenus (moyenne de 77 UI/dl) à 35 semaines48. Un suivi à long terme actualisé sur ces patients a révélé après trois ans un bienfait pertinent sur le plan clinique avec une réduction conséquence des taux de saignement annualisé (TSA de 0 pour les deux concentrations de vecteur) et une activité moyenne du FVIII de 20 UI/dl dans le groupe recevant la dose élevée et de 13 UI/dl, dans le groupe recevant la dose moins importante49.
- Les données intermédiaires de l’étude de phase III de Biomarin sur 132 patients recevant une dose élevée de vecteur (6 x 1013 vg/kg) a montré une concentration moyenne de FVIII de 41,9 UI/dl (valeur médiane : 24,4 UI/dl) après un an et de 22,9 UI/dl (valeur médiane : 14,7 UI/dl) après deux ans50. Cette baisse de l’expression du FVIII au fil du temps, également observée dans les études de phase précoce sur un plus petit nombre de patients, jette un doute quant à la durabilité à long terme de cette thérapie génique axée sur le FVIII, et impose d’autres examens ainsi qu’une surveillance.
- Une étude clinique de phase III porte sur le AVV-Spark200 (SPK-8011, Roche/Spark) qui utilise un vecteur généré à partir d’une bibliothèque de capsides après sélection pour le tropisme d’hépatocytes humains. Des données préliminaires de l’essai montrent une activité du FVIII entre 10 et 13 % après l’administration de doses faibles et modérées du vecteur.
- Une étude de Pfizer (Sangamo) utilisant le vecteur viral adéno-associé 2/6 recombinant codant pour le FVIII humain est en phase III.
De nombreux vecteurs de thérapie génique sont actuellement examinés dans des études cliniques de phase III et certains ont d’ores et déjà été soumis pour une évaluation initiale en vue d’une approbation de la FDA (Food and Drug Administration). Il est probable que la thérapie génique constituera une option thérapeutique offerte pour l’hémophilie dans un à deux ans.51
e) Traitement de la maladie de von Willebrand52-54
I. Desmopressine (DDAVP)
- La plupart des patients accusant un léger déficit quantitatif en facteur de von Willebrand (FvW) (maladie de von Willebrand de type 1) et certains patients présentant des anomalies qualitatives de ce facteur (maladie de von Willebrand de types 2A et 2M) répondent à la desmopressine (par voie intraveineuse ou sous-cutanée à raison de 0,3 mg/kg de masse corporelle).
- La desmopressine par voie intranasale (à raison de 150 mg si la masse corporelle est égale ou inférieure à 50 kg et de 2 x 150 mg si elle supérieure à 50 kg), bien qu’efficace, n’est pas offerte au Canada à l’heure actuelle.
- On conseille d’effectuer au préalable des tests vérifiant la réponse à la desmopressine afin de confirmer qu’elle corrige bien les taux de FvW pour traiter les épisodes hémorragiques mineurs ou majeurs. Les patients atteints de la maladie de type 3 (absence quasi totale de FvW), de même que certains patients souffrant de la maladie de type 2A ou de type 2M, ne répondent pas à la desmopressine.
- L’administration de la desmopressine peut provoquer une thrombocytopénie chez les personnes atteintes de la maladie de von Willebrand de type 2B; elle n’est donc généralement pas recommandée chez ces patients.
- Dans les atteintes de type 1C (type Vicenza) et de type 2N, la réponse de pointe au traitement pourrait être normale, mais la demi-vie des facteurs de coagulation augmentés (FVIII ou FvW dans le type 1C; FVIII dans le type 2N) sera considérablement écourtée.
II. Concentrés de facteurs de coagulation
- En l’absence de réponse à la desmopressine et en cas d’hémorragie grave ou d’interventions majeures, on peut administrer du FVIII/FvW dérivé de plasma (Humate-P, Wilate) ou du concentré de FvW recombinant (Vonvendi, approuvé par Santé Canada, mais non encore offert par la Société canadienne du sang ni Héma-Québec au moment de la rédaction du présent chapitre).
- Les concentrés de FVIII et de FvW contiennent ces facteurs de coagulation dans diverses proportions selon le produit, mais le concentré de FvW recombinant ne renferme pas de FVIII (voir le tableau 1 du chapitre 5 du présent Guide).
- La dose habituelle est de 30 à 50 unités par kg (unités de cofacteurs de la ristocétine pour le produit Humate-P ou unités de FVIII pour le produit Wilate) dans le cas d’hémorragies légères, et de 50 à 80 unités par kg dans le cas d’hémorragies plus graves. Les patients atteint de la maladie de von Willebrand des types 2 et 3 doivent recevoir la dose la plus élevée de la gamme posologique. Une dose supplémentaire peut être administrée toutes les 12 heures, selon le contexte clinique.
- Chez les patients réfractaires aux concentrés de FVIII ou de FvW, on peut ajouter l’administration de desmopressine ou la transfusion de plaquettes.
- Bien que le FvW soit nécessaire à la cessation initiale du saignement d’une muqueuse, l’administration de taux adéquats de FVIII est importante pour l’arrêt d’une hémorragie des tissus mous et d’une hémorragie consécutive à une intervention chirurgicale ainsi que pour le maintien de l’hémostase (après l’hémostase primaire avec la formation du clou plaquettaire).
- Lorsque ces concentrés doivent être utilisés pendant une période prolongée, on conseille de surveiller la concentration de FVIII et de maintenir son activité en dessous de 200 UI/dl (200 %) afin de diminuer les risques d’effets thrombogènes, surtout chez les personnes ayant subi une intervention chirurgicale et les patients immobilisés.
- Puisque le concentré de FvW recombinant ne contient pas de FVIII, on s’attendra à une période de latence avant que le taux de FVIII in vivo n’augmente par suite d’une perfusion de FvW recombinant. Ainsi, pour les interventions chirurgicales chez les patients atteints d’une forme grave de la maladie de von Willebrand (p. ex. de type 3 et de type 1 grave) sans traitement préventif par le FvW, il faut administrer du FVIII recombinant en même temps que la dose initiale de FvW recombinant immédiatement avant l’intervention, ou administrer une dose d’attaque de FvW recombinant quelques heures avant l’incision afin de permettre au taux de FVIII endogène d’atteindre la concentration voulue avant l’intervention.
f) Troubles de la coagulation congénitaux rares55,56
Certains patients présentent un déficit de facteur de coagulation congénital rare avec diathèse hémorragique. Il s’agit notamment des déficits en FII, FV, FVII, FX, FXI, fibrinogène et FXIII, dont l’incidence respective est de 1 sur 500 000 et de 1 sur 2 000 000 dans la population. On trouvera au tableau 2 un résumé du traitement des hémorragies chez les patients présentant un déficit d’un de ces facteurs de coagulation. Les caractéristiques des concentrés de facteurs de coagulation utilisés pour remédier aux déficits sont résumées au tableau 1 du chapitre 5 du présent Guide.
Tableau 2 : Traitement d’un patient présentant un déficit de facteur de coagulation rare.
Remarque : Pour plus de précisions sur les concentrés de facteurs de coagulation offerts, consulter le tableau 1 au chapitre 5 du présent Guide.
| Déficit | Demi-vie plasmatique |
Récupération in vivo# (UI/dl¶ par UI/kg, sauf pour le fibrinogène) |
Concentrations désirées¶ | Options thérapeutiques | Observations |
|---|---|---|---|---|---|
| Fibrinogène | De 2 à 4 jours |
0,017-0,018 g/l par mg/kg perfusé (Une perfusion de 10 mg/kg se traduit par une hausse de ~ 0,2 g/l du taux sanguin de fibrinogène) |
|
|
|
| FII | De 2 à 3 jours | ~1,0 |
|
|
|
| FV | De 15 à 36 heures | ~1,6 |
|
|
|
| FVII | De 3 à 6 heures | ~2,0 |
|
|
|
| FX | De 20 à 40 heures | 1–1,9 |
|
|
|
| FXI | De 35 à 60 heures | ~1,8 |
|
|
|
| FXIII | De 5 à 11 jours | 1,0–2,0 |
|
|
|
|
¶ 1 UI/dl = activité de 1 % (= 0,01 UI/ml) # On peut s’attendre à ce que la récupération in vivo (RIV) de chaque produit varie d’une personne à l’autre. † Plasma : plasma conservé/décongelé, plasma frais congelé/plasma-aphérèse frais congelé ou plasma viro-inactivé par un traitement par solvant/détergent (p. ex. Octaplasma [Octapharma]). Si l’administration de plasma ne permet pas d’atteindre la concentration désirée et l’hémostase, il faut parfois procéder à une aphérèse avec remplacement plasmatique. * Si l’administration de plasma frais congelé/plasma-aphérèse frais-congelé ou de plasma viro-inactivé par un traitement par solvant/détergent (p. ex. Octaplasma [Octapharma]) ne permet pas d’atteindre la concentration désirée et l’hémostase, il faut parfois procéder à une aphérèse avec remplacement du plasma frais congelé. ‡ CCP : concentré de complexe prothrombique. Risque de thrombose – administrer la dose minimale efficace. |
|||||
2. Anomalies plaquettaires congénitales57-60
Il existe de nombreux types d’anomalies fonctionnelles plaquettaires congénitales. L’Association canadienne des directeurs de cliniques d’hémophilie (ACDCH) a publié une liste des anomalies fonctionnelles plaquettaires et de leurs critères diagnostiques61 ainsi que l’algorithme d’analyse62. Ces troubles comprennent, entre autres, la thrombasthénie de Glanzmann (déficit ou anomalie fonctionnelle de la glycoprotéine IIb/IIIa [intégrine αIIbβ3] de la membrane plaquettaire), le syndrome de Bernard-Soulier (déficit ou anomalie fonctionnelle de la glycoprotéine Ib, IX ou V) et le syndrome du pool vide. La thrombasthénie de Glanzmann a une incidence globale faible à l’échelle internationale (1 personne sur 1 million), mais bien supérieure (jusqu’à 1 personne sur 40 000-100 000) dans les zones où prévalent les unions consanguines63,64.
a) Traitement des hémorragies légères/modérées
- La plupart des hémorragies légères/modérées chez les patients atteints de ces affections peuvent être maîtrisées par des mesures conservatrices, dont l’application locale de pression, l’administration d’antifibrinolytiques et l’emploi d’hémostatiques topiques, tels que la colle de fibrine.
- La desmopressine (DDAVP) peut également être efficace en présence d’une hémorragie légère ou modérée, mais la réponse à cet agent varie d’un patient à un autre59.
b) Traitement des hémorragies graves
- Les hémorragies graves qui ne répondent pas aux mesures conservatrices peuvent être gérées par des transfusions de plaquettes (idéalement d’aphérèse, dont les antigènes leucocytaires humains [HLA] sont compatibles; néanmoins, il ne faut pas retarder le traitement pour des questions de disponibilité en cas de besoin urgent). Au Canada, les concentrés plaquettaires offerts sont déleucocytés (ce qui réduit l’allo-immunisation anti-HLA, voir le chapitre 18).
- Chez les patients transfusés qui ont développé des anticorps anti-HLA ou des anticorps dirigés contre les glycoprotéines plaquettaires manquantes et qui sont réfractaires aux transfusions de plaquettes, le rFVIIa apparaît utile selon des registres et études de cas.
- D’après les résultats obtenus chez les patients présentant une thrombasthénie de Glanzmann, l’administration de rFVIIa à raison d’environ 90 µg/kg toutes les 2 à 2,5 heures (deux ou trois doses ou plus, s’il y a lieu), en association avec des antifibrinolytiques, est efficace dans une forte proportion d’hémorragies et d’interventions chirurgicales :
- chez les adultes et les enfants, quel que soit le statut d’anticorps plaquettaires59,63,65,66. Dans la mesure du possible, il est préférable d’éviter les transfusions de plaquettes chez des femmes en âge de procréer et chez des filles prépubères atteintes d’une thrombasthénie de Glanzmann. Pendant la grossesse; des allo-anticorps dirigés contre le complexe GPIIb/IIIa plaquettaire (intégrine αIIbβ3) peuvent traverser la barrière placentaire et être néfastes pour le fœtus et le nouveau-né (thrombocytopénie, saignement). On note également un problème similaire pour les anticorps anti-GPIb-IX en cas de syndrome de Bernard-Soulier.
- Selon les résultats peu nombreux dont on dispose au sujet de la perfusion continue de rFVIIa, il semble que cette approche ne soit pas aussi efficace pour arrêter une hémorragie, mais qu’elle le soit à titre prophylactique lors d’une intervention chirurgicale63,67. On a fait état de complications thrombotiques lors de la perfusion continue de rFVIIa à fortes doses pendant une période prolongée à des opérés présentant d’autres facteurs de risque de thrombose68.
3. Troubles vasculaires/du tissu conjonctif
Les hémorragies graves sont rares chez les patients atteints de troubles vasculaires/du tissu conjonctif et, dans la plupart des cas, elles peuvent être maîtrisées par des mesures conservatrices. La desmopressine a été employée avec succès chez certaines personnes subissant une intervention chirurgicale, probablement parce qu’elle améliore l’interaction entre les plaquettes et l’endothélium. En cas de saignement d’une muqueuse, on peut également utiliser de l’acide tranexamique. On ne dispose pas de données montrant que les produits sanguins sont indiqués chez ces patients.
Coagulopathies acquises
1. Maladie du foie
À l’exception du facteur tissulaire, tous les facteurs de coagulation sont synthétisés dans le foie; certains d’entre eux (FII, FVII, FIX et FX) ont besoin de la participation de la vitamine K, qui agit à titre de cofacteur.
Chez les patients atteints d’une maladie du foie, le taux de facteurs de coagulation est souvent faible, sauf ceux du fibrinogène et du FVIII, qui sont des réactifs de phase aiguë et dont la concentration tend à augmenter en cas de maladie du foie non compliquée. Une coagulation intravasculaire disséminée concomitante/coagulopathie de consommation concomitante doit être envisagée en présence d’une diminution des taux de fibrinogène et de FVIII.
Le temps de céphaline activée et le temps de prothrombine3 sont habituellement allongés dans les cas de maladie du foie et suffisent généralement à la surveillance du traitement sans que l’on doive procéder à des épreuves pour déterminer les taux de facteurs de coagulation. Il convient de noter que le rapport international normalisé (RIN) a été validé précisément (et uniquement) pour le suivi thérapeutique de l’anticoagulation orale par des antagonistes de la vitamine K (p. ex. warfarine). Il n’a pas été validé pour les anomalies de coagulation associées à des maladies hépatiques (ou autres) et n’a pas de corrélation avec le risque de saignement chez des patients atteints d’une affection hépatique. Bien que la plupart des facteurs de coagulation soient fabriqués dans le foie et que leurs taux diminuent généralement en cas de dysfonctionnement hépatocellulaire, on observe une hausse des taux de fibrinogène (et de FVIII) en tant que réactifs de phase aiguë (voir la figure sur les changements dans l’hémostase en cas de coagulation intravasculaire disséminée (CIVD) et de maladie hépatique dans l’ouvrage de révision illustré de Elbaz et Sholzberg3). Par conséquent, un taux de fibrinogène normal pourrait être trop bas en cas de maladie hépatique et le patient doit faire l’objet d’une surveillance afin de détecter une coagulation intravasculaire disséminée à titre de complication d’une affection hépatocellulaire. Les personnes atteintes d’une maladie du foie peuvent aussi présenter une thrombocytopénie en raison d’une splénomégalie causée par une hypertension portale ou une infection virale sous-jacente.
Les saignements résultant d’une coagulopathie liée à une maladie du foie sont en général légers et peuvent habituellement être maîtrisés adéquatement par une perfusion de plasma, qui contient tous les facteurs de coagulation synthétisés dans le foie. Le concentré de complexe prothrombique est thrombogène dans la maladie du foie et doit de préférence être évité chez les patients qui en sont atteints69. Même si on a eu recours aux concentrés de complexe prothrombique à la place du plasma pour éviter la surcharge volémique dans ce contexte, on dispose uniquement de données probantes limitées concernant l’efficacité et l’innocuité pour étayer cette pratique69. Les hémorragies découlant de lésions structurales, telles que les varices et les ulcères, peuvent être importantes chez ces patients; on doit ici tenter de parvenir à l’hémostase au siège de l’hémorragie tout en traitant la coagulopathie.
2. Surdose d’anticoagulant par voie orale
a) Antagonistes de la vitamine K (AVK)70,71
La vitamine K est nécessaire à la synthèse des facteurs de coagulation fonctionnels FII, FVII, FIX et FX (facteurs dépendants de la vitamine K). Les médicaments coumariniques produisent leur effet anticoagulant en inhibant de façon compétitive la vitamine K, ce qui provoque une diminution de la fonction des facteurs dépendants de la vitamine K et une augmentation du ratio international normalisé (temps de prothrombine).
De nombreux médicaments peuvent interagir avec la coumarine et entraîner une anticoagulation excessive associée à des augmentations marquées du ratio international normalisé. Lorsque le rapport international normalisé est modérément augmenté sans qu’il y ait d’hémorragie, il peut suffire de cesser l’utilisation de l’anticoagulant pour permettre l’augmentation progressive des facteurs dépendants de la vitamine K, selon leur taux de synthèse individuel.
Lorsqu’il faut augmenter immédiatement le taux des facteurs de coagulation, par exemple, en présence d’une hémorragie aiguë ou lorsqu’une intervention chirurgicale d’urgence s’impose, l’administration d’un concentré de complexe prothrombique (Octaplex ou Beriplex) par perfusion (pour obtenir les directives posologiques, consulter la version de 2022 des Recommandations concernant l’utilisation des concentrés de complexe prothrombique au Canada du Comité consultatif national sur le sang et les produits sanguins72) en association avec la vitamine K permet d’obtenir une réponse soutenue. (Se reporter également à la rubrique sur les concentrés de complexe prothrombique dans le tableau 4 du chapitre 5 du présent Guide.) Dans le cas d’une intervention chirurgicale non urgente, il est généralement suffisant d’interrompre la prise de coumarine (remplacée par une héparine de bas poids moléculaire, s’il y a lieu) durant les jours précédant l’opération pour permettre de ramener le ratio international normalisé en dessous de 1,5.
Chez les patients qui font une hémorragie lorsqu’ils prennent des antagonistes de la vitamine K, il faut déterminer la source de l’hémorragie et traiter le saignement une fois pour toutes.
b) Anticoagulants oraux directs (AOD, inhibiteurs oraux du FXa et du FIIa)70,71,73,74
Les anticoagulants oraux directs offerts au Canada sont les inhibiteurs directs du FIIa (dabigatran) et du FXa (rivaroxaban, apixaban et edoxaban). Tous les anticoagulants oraux directs dépendent en partie de l’élimination rénale aux fins de clairance de la circulation; le dabigatran en est le plus dépendant, soit dans une proportion de 85 %. Pendant la prise de ces anticoagulants, il est important de surveiller la fonction rénale, car il peut y avoir bioaccumulation en cas d’insuffisance rénale. Les anticoagulants oraux directs ne nécessitent pas une surveillance systématique de la coagulation en laboratoire ni un ajustement de la dose. Toutefois, il est important d’évaluer leur présence et leur concentration chez les patients sous anticoagulant qui présentent une hémorragie nécessitant de toute urgence une intervention chirurgicale ou une thrombolyse (voir la figure sur l’effet des AOD sur les épreuves hémostatiques dans Elbaz et Sholzberg)3.
I. Dabigatran
Un temps de thrombine normal exclut essentiellement la présence de dabigatran. Il convient de noter qu’un temps de thrombine prolongé ne permet pas de faire la distinction entre des taux de dabigatran cliniquement importants et non significatifs (le test est très sensible) et qu’un temps de céphaline activée normal n’exclut pas la présence de dabigatran circulant (le test peut être insensible au dabigatran selon les réactifs utilisés). Le temps de thrombine diluée, qui convient le mieux pour évaluer avec précision l’activité anticoagulante du dabigatran, n’est pas largement accessible.
II. Inhibiteurs du FXa
Pour les inhibiteurs directs du FXa (rivaroxaban, apixaban et edoxaban), le temps de prothrombine est généralement prolongé, mais un temps de prothrombine normal n’exclut pas des taux cliniquement pertinents d’un inhibiteur direct du FXa (le test peut être insensible selon les réactifs utilisés). Une activité normale anti-Xa exclut les taux cliniquement pertinents d’inhibiteurs directs du FXa, mais elle est utile pour quantifier les taux plasmatiques d’un médicament uniquement quand elle est calibrée avec le médicament en question.
Le traitement des hémorragies chez les patients sous anticoagulants oraux directs est complexe. Veuillez vous reporter aux voies ou aux algorithmes de traitement locaux et aux lignes directrices publiées70,73. Thrombose Canada publie en ligne des guides cliniques qui sont régulièrement mis à jour74.
- Pour les saignements légers, une thérapie locale ou l’omission de la ou des prochaines doses d’anticoagulants oraux directs peut suffire.
- En cas d’hémorragies cliniquement significatives ou mettant en danger la vie du patient et d’hémorragies ne réagissant pas aux mesures de soutien locales et générales (y compris la transfusion), les mesures suivantes sont proposées :
- Administrer des antidotes propres aux anticoagulants oraux directs :
- idarucizumab (Parxbind, un fragment d’anticorps monoclonal, homologué au Canada) pour le dabigatran;
- andexanet alfa (AndexXa, un anticorps recombinant humain modifié du FXa, approuvé par la FDA aux États-Unis, mais non encore homologué au Canada) pour les inhibiteurs du FXa.
- Envisager l’hémodialyse pour réduire le taux de médicament circulant chez les patients sous dabigatran qui souffrent d’une hémorragie mettant leur vie en danger, d’une insuffisance rénale ou d’un temps de céphaline activée excessivement prolongé (ou dont le taux de dabigatran est supérieur à 500 ng/ml) et quand on ne dispose pas d’idarucizumab. L’hémodialyse ne convient pas aux patients sous rivaroxaban et apixaban, car ces médicaments sont liés aux protéines, et donc non dialysables.
- Des études de cohortes prospectives ont également montré l’utilité d’un concentré de complexe prothrombique activé (FEIBA®, 50 UI/kg, 2 000 UI maximum)72,75 pour le dabigatran et l’utilisation de concentrés de complexe prothrombique régulier (CCP : Octaplex, Beriplex, 25‑50 UI/kg, 3 000 UI maximum) pour les inhibiteurs directs du FXa72,76. Les bienfaits et les risques cliniques qui y sont associés (p. ex. thrombose) devront être précisés dans le cadre d’études cliniques.
- Les antifibrinolytiques (acide tranexamique) peuvent également être utilisés en cas d’hémorragie, mais ils ne doivent pas être utilisés de façon concomitante avec un concentré de complexe prothrombique activé ou un concentré de complexe prothrombique (ou en cas d’hématurie macroscopique ou d’hémorragie dans la cavité thoracique; voir les figures sur les effets secondaires de l’acide tranexamique systémique et les contre-indications de l’acide tranexamique dans Relke et al.5).
- Pour le traitement peropératoire d’une intervention chirurgicale urgente ou non chez des patients prenant des anticoagulants oraux directs, consultez les documents de référence70,73.
3. Coagulation intravasculaire disséminée (CIVD)77
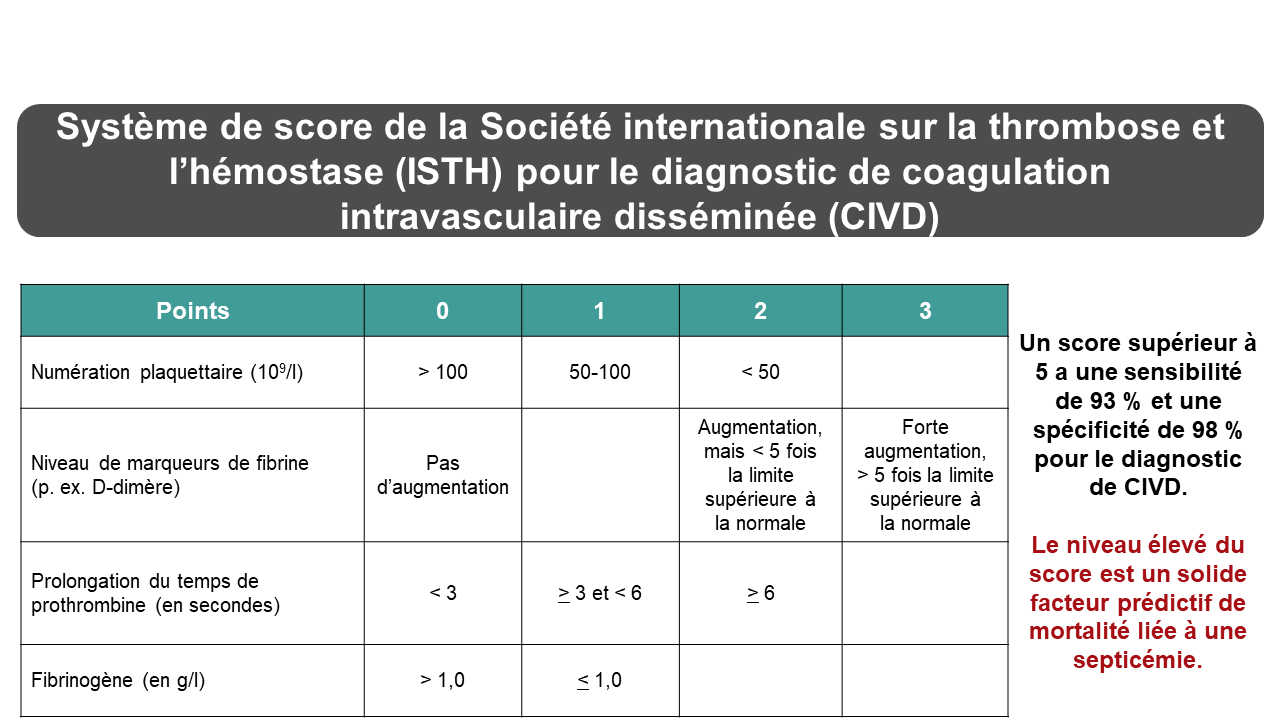
La coagulation intravasculaire disséminée peut être déclenchée par plusieurs situations cliniques, dont une destruction massive de tissus, une infection, des complications obstétriques et un cancer. Une activation non contrôlée du système de coagulation va entraîner l’activation ainsi que la consommation de facteurs de coagulation et de plaquettes. De plus, une importante activation fibrinolytique secondaire se traduira par la destruction de facteurs de coagulation et par la formation de produits de dégradation de la fibrine et du fibrinogène nuisant à la polymérisation de la fibrine et à la fonction plaquettaire. Il existe plusieurs systèmes de score validés pour le diagnostic d’une CIVD. La figure 2 présente le système de score de la Société internationale sur la thrombose et l’hémostase pour une CIVD déclarée78 (total de 8 points, ≥ 5 points = CIVD) s’appuyant sur des épreuves de laboratoire simples largement disponibles3. La figure montre également les bonnes sensibilité et spécificité du diagnostic de CIVD de ce système et la valeur prédictive de scores élevés en termes de mortalité79. Selon chaque patient, la valeur prédictive positive (d’un score ≥ 5) indiquant la présence d’une CIVD était de 96 % et la valeur prédictive négative (d’un score < 5) indiquant l’absence d’une CIVD était de 97 %79. Selon l’équilibre entre la coagulation et la fibrinolyse, les patients peuvent présenter des complications hémorragiques ou thrombotiques. Chez un patient n’ayant pas d’hémorragie, l’administration d’un traitement visant à supprimer le stimulus déclenchant la coagulation intravasculaire disséminée est très importante et suffit souvent à inverser le processus. Lorsque survient une hémorragie, on peut stabiliser l’état du patient en remplaçant les facteurs de coagulation consommés. Le traitement peut inclure la transfusion de plasma, de concentré de fibrinogène, de cryoprécipité (pour le FVIII et le fibrinogène) et de plaquettes (en cas de thrombocytopénie grave). Le concentré de complexe prothrombique est thrombogène en cas de coagulation intravasculaire disséminée et est contre-indiqué69. La transfusion est uniquement réalisée en appoint au traitement du problème clinique sous-jacent ayant provoqué la coagulation intravasculaire disséminée. Le remplacement des facteurs de coagulation n’arrête pas le processus de coagulation intravasculaire disséminée.
Une septicémie bactérienne foudroyante avec coagulation intravasculaire disséminée et nécrose cutanée est associée à un fort taux de morbidité et de mortalité, et on a évalué la place éventuelle des inhibiteurs naturels de protéases en tant qu’agents thérapeutiques. Les résultats d’études cliniques de phase III semblent indiquer au départ que l’administration de protéine C activée recombinante améliore la survie des personnes atteintes80. Cette constatation n’a toutefois pas été confirmée dans les études subséquentes, notamment une vaste étude comparative avec placebo chez des patients souffrant d’une septicémie grave et d’un choc septique81. Une vaste étude clinique de phase III portant sur l’administration d’antithrombine n’a pas non plus montré de bienfaits pour la survie82, quoique de récentes études rétrospectives ajustées en fonction de la propension menées au Japon aient montré un important bienfait chez les patients souffrant d’une septicémie grave avec coagulation intravasculaire disséminée sous anti-thrombine83. Ces dernières observations nécessiteront une validation prospective. La thrombomoduline soluble recombinante est un autre agent prometteur selon une étude rétrospective effectuée dans plusieurs centres84.
Angiœdème héréditaire
Angiœdème héréditaire85
L’angiœdème héréditaire est dû à un déficit quantitatif ou fonctionnel en inhibiteur de la C1 estérase (C1-INH), qui joue un rôle régulateur clé dans le système du complément, le système de coagulation intrinsèque et le système fibrinolytique. La prévalence de l’angiœdème héréditaire est d’environ 1 sur 50 000. Ce trouble est associé à des mutations du gène SERPING1 (autrefois connu sous le nom de C1-INH), situé sur le chromosome 11. Il s’agit d’une maladie héréditaire à transmission autosomique dominante.
Deux types principaux d’angiœdème héréditaire ont été décrits : l’angiœdème héréditaire de type 1 est dû à un déficit quantitatif en antigène C1-INH circulant (85 % des cas); et l’angiœdème héréditaire de type 2 est associé à un taux normal d’antigène C1-INH, mais il présente un déficit fonctionnel (15 % des cas)86. Un troisième type d’angiœdème héréditaire a également été décrit, mais il n’est associé à aucun déficit en C1-INH. Les symptômes cliniques sont attribués à une production excessive de bradykinine87.
Les patients présentant un angiœdème héréditaire souffrent de gonflements épisodiques, appelés « crises », qui peuvent toucher n’importe quelle partie du corps. Les crises d’angiœdème héréditaire affectent souvent la peau, le visage, les voies respiratoires supérieures, l’oropharynx, le tractus gastro-intestinal, l’extrémité des membres et les organes génitaux. Les œdèmes du visage et de l’oropharynx demeurent les plus préoccupants, car ils peuvent être associés à une obstruction des voies aériennes potentiellement mortelle. L’œdème du tractus gastro-intestinal est souvent associé à des douleurs intenses, à des nausées, à des vomissements, à de la diarrhée et à une occlusion intestinale temporaire pouvant donner lieu à des interventions chirurgicales inutiles. Les déclencheurs courants des gonflements sont, entre autres, le stress, des médicaments, les traumatismes, une infection ou une exposition à des hormones.
Bien qu’il soit souvent confondu avec l’angiœdème allergique ou anaphylactique, l’angiœdème héréditaire se distingue de ces maladies par l’absence d’urticaire et la faible progression des symptômes. Un épisode de gonflement aigu entraîne généralement un œdème progressif du site affecté pendant 24 heures, suivi d’une résorption graduelle sur une période d’un à cinq jours en l’absence de traitement. Toutefois, la fréquence, la durée et la gravité des crises sont variables d’une personne à l’autre. L’angiœdème héréditaire ne doit pas être considéré comme une maladie bénigne puisqu’on observe un taux de mortalité atteignant 30 % chez les personnes non traitées ou pour lesquelles un mauvais diagnostic avait été posé88.
La prise en charge de l’angiœdème héréditaire repose généralement sur deux stratégies : le traitement du gonflement aigu (sur demande) et le traitement pour prévenir les épisodes de gonflement (préventif). Le traitement sur demande peut consister à remplacer le C1-INH ou à réduire la production ou le rôle de la bradykinine. Il est recommandé aux patients ayant un angiœdème héréditaire d’avoir toujours en leur possession une carte pour portefeuille (HAE _ AOH Carte pour portefeuille – version anglaise; HAE _ AOH Carte pour portefeuille – version française) les identifiant afin d’aider le personnel médical en cas d’urgence. Après le traitement initial d’urgence, le centre hospitalier ou le(s) médecin(s) responsable(s) du patient doivent être informés en vue d’obtenir les recommandations concernant les soins continus. L’établissement/le(s) médecin(s) doivent également être consultés pour connaître les recommandations en matière de couverture hémostatique pour tout acte chirurgical ou effractif.
1. Options thérapeutiques pour le traitement sur demande
- Produits de remplacement du C1-INH (inhibiteur de la C1 estérase) dérivé du plasma, comme Berinert, à raison de 20 U/kg par voie intraveineuse
- Icatibant (Firazyr), antagoniste des récepteurs B2 de la bradykinine, à raison de 30 mg par injection sous-cutanée lente toutes les 6 heures, jusqu’à concurrence de trois doses en 24 heures
- Plasma, à raison de 10 à 15 ml/kg toutes les deux à quatre heures jusqu’à observation d’une amélioration clinique
REMARQUE : Les corticostéroïdes, l’épinéphrine et les antihistaminiques sont inefficaces pour traiter l’angiœdème héréditaire.
2. Options thérapeutiques pour le traitement préventif
- Remplacement du C1-INH dérivé du plasma : Berinert, à raison de 20 U/kg par voie intraveineuse deux fois par semaine, ou Cinryze à raison de 1 000 U par voie intraveineuse deux fois par semaine
- Concentré de C1-INH dérivé du plasma par voie sous-cutanée : HAEGARDA 60 UI/kg deux fois par jour
- Lanadélumab (Takhzyro), anticorps monoclonal inhibant la kallikréine plasmatique, à raison de 300 mg par voie sous-cutanée toutes les deux semaines. Un intervalle de quatre semaines entre les administrations de 300 mg peut être envisagé une fois que l’état du patient est sous contrôle.
- Dans des cas particuliers, on peut envisager l’acide tranexamique oral (25 mg/kg administrés tous les jours en doses fractionnées) ou le danazol (de 200 à 600 mg tous les jours).
Crédits de développement professionnel continu
Les associés et les professionnels de la santé qui participent au Programme de maintien du certificat du Collège royal des médecins et chirurgiens du Canada peuvent demander que la lecture du Guide de la pratique transfusionnelle soit reconnue comme activité de développement professionnel continu au titre de la Section 2 — Apprentissage individuel. Ces personnes peuvent réclamer 0,5 crédit par heure de lecture, jusqu’à hauteur de 30 crédits par année.
Les technologistes médicaux qui participent au Programme d’enrichissement professionnel (PEP) de la Société canadienne de science de laboratoire médical peuvent demander que la lecture du Guide de la pratique transfusionnelle soit reconnue en tant qu’activité non vérifiée
Remerciements
Les auteurs remercient Kathryn Webert, M.D., M. Sc., FRCPC, et Aditi Khandelwal, MDCM, FRCPC, qui ont révisé le présent chapitre.
Suggestion de citation
Poon M, Goodyear MD, Rydz N and Lee A. Troubles de l’hémostase et angiœdème héréditaire. Dans : Clarke G, Abe T (dir.). Guide de la pratique transfusionnelle, [Internet]. Ottawa, Société canadienne du sang. 2022 [cité le AAAA MM JJ]. Chapitre 17. Disponible sur le Web : https://developpementprofessionnel.sang.ca
N’hésitez pas à nous faire part de vos suggestions et de vos questions concernant le Guide de la pratique transfusionnelle par le biais de notre formulaire.
Bibliographie
- World Federation of Hemophilia. Compendium of Assessment Tools, World Federation of Hemophilia 2014. https://elearning.wfh.org/resource/compendium-of-assessment-tools/ (Last accessed June 13, 2022)
- James P. Let's Talk Period, Self-Bat. Kingston, Ontario, Queen's University. https://letstalkperiod.ca/self-bat/ (Last accessed May 4, 2022)
- Elbaz C, Sholzberg M. An Illustrated Review of Bleeding Assessment Tools and Common Coagulation Tests. Research and Practice in Thrombosis and Haemostasis 2020; 4: 761-73. https://onlinelibrary.wiley.com/doi/abs/10.1002/rth2.12339.
- Sharma R, Haberichter SL. New Advances in the Diagnosis of Von Willebrand Disease. Hematology 2019; 2019: 596-600. https://doi.org/10.1182/hematology.2019000064.
- Relke N, Chornenki NLJ, Sholzberg M. Tranexamic Acid Evidence and Controversies: An Illustrated Review. Research and Practice in Thrombosis and Haemostasis 2021; 5: e12546. https://onlinelibrary.wiley.com/doi/abs/10.1002/rth2.12546.
- Canadian Hemophilia Society. Treatment Centres. https://www.hemophilia.ca/find-a-treatment-center-near-you/ (Last accessed June 25, 2018)
- Srivastava A, Santagostino E, Dougall A, Kitchen S, Sutherland M, Pipe SW, Carcao M, Mahlangu J, Ragni MV, Windyga J, Llinás A, Goddard NJ, Mohan R, Poonnoose PM, Feldman BM, Lewis SZ, van den Berg HM, Pierce GF, on behalf of the WFH Guidelines for the Management of Hemophilia panelists and co-authors. Wfh Guidelines for the Management of Hemophilia, 3rd Edition. Haemophilia 2020; 26: 1-158. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.14046.
- Hews-Girard J, Rydz N, Lee A, Goodyear MD, Poon M-C. Desmopressin in Non-Severe Haemophilia A: Test-Response and Clinical Outcomes in a Single Canadian Centre Review. Haemophilia 2018; 24: 720-5. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.13586.
- Janneke IL, Marieke JHAK, Manuel C, Shannon J, Alice SvV, Marjolein P, Elena S, Helen P, Erik B, Jan V, Johanna GvdB, Karin F, for the Rc. Desmopressin in Moderate Hemophilia a Patients: A Treatment Worth Considering. Haematologica 2018; 103: 550-7. https://haematologica.org/article/view/8393.
- Revel-Vilk S, Blanchette VS, Sparling C, Stain AM, Carcao MD. DDAVP Challenge Tests in Boys with Mild/Moderate Haemophilia a*. Br J Haematol 2002; 117: 947-51. https://onlinelibrary.wiley.com/doi/abs/10.1046/j.1365-2141.2002.03507.x.
- The Web-Accessible Population Pharmacokinetic Service - Hemophilia (Wapps-Hemo) Project, May 16, 2018 2018. https://www.wapps-hemo.org/default.aspx (Last accessed June 25, 2018.
- Manco-Johnson MJ, Abshire TC, Shapiro AD, Riske B, Hacker MR, Kilcoyne R, Ingram JD, Manco-Johnson ml, Funk S, Jacobson L, Valentino LA, Hoots WK, Buchanan GR, DiMichele D, Recht M, Brown D, Leissinger C, Bleak S, Cohen A, Mathew P, Matsunaga A, Medeiros D, Nugent D, Thomas GA, Thompson AA, McRedmond K, Soucie JM, Austin H, Evatt BL. Prophylaxis Versus Episodic Treatment to Prevent Joint Disease in Boys with Severe Hemophilia. N Engl J Med 2007; 357: 535-44. http://www.ncbi.nlm.nih.gov/pubmed/17687129.
- Feldman BM, Rivard GE, Babyn P, Wu JKM, Steele M, Poon MC, Card RT, Israels SJ, Laferriere N, Gill K, Chan AK, Carcao M, Klaassen RJ, Cloutier S, Price VE, Dover S, Blanchette VS. Tailored Frequency-Escalated Primary Prophylaxis for Severe Haemophilia A: Results of the 16-Year Canadian Hemophilia Prophylaxis Study Longitudinal Cohort. Lancet Haematol 2018. https://www.ncbi.nlm.nih.gov/pubmed/29731369.
- Den UIjl IEM, Fischer K, Van Der Bom JG, Grobbee DE, Rosendaal FR, Plug I. Analysis of Low Frequency Bleeding Data: The Association of Joint Bleeds According to Baseline FVIII Activity Levels. Haemophilia 2011; 17: 41-4. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1365-2516.2010.02383.x.
- Fischer K, Steen Carlsson K, Petrini P, Holmström M, Ljung R, van den Berg HM, Berntorp E. Intermediate-Dose Versus High-Dose Prophylaxis for Severe Hemophilia: Comparing Outcome and Costs since the 1970s. Blood 2013; 122: 1129-36. https://doi.org/10.1182/blood-2012-12-470898.
- WU R, LUKE K-H, POON M-C, WU X, ZHANG N, ZHAO L, SU Y, ZHANG J. Low Dose Secondary Prophylaxis Reduces Joint Bleeding in Severe and Moderate Haemophilic Children: A Pilot Study in China. Haemophilia 2011; 17: 70-4. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1365-2516.2010.02348.x.
- Verma SP, Dutta TK, Mahadevan S, Nalini P, Basu D, Biswal N, Ramesh A, Charles D, Vinod KV, Harichandra Kumar KT. A Randomized Study of Very Low-Dose Factor VIII Prophylaxis in Severe Haemophilia – a Success Story from a Resource Limited Country. Haemophilia 2016; 22: 342-8. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.12838.
- Mahlangu J, Oldenburg J, Paz-Priel I, Negrier C, Niggli M, Mancuso ME, Schmitt C, Jiménez-Yuste V, Kempton C, Dhalluin C, Callaghan MU, Bujan W, Shima M, Adamkewicz JI, Asikanius E, Levy GG, Kruse-Jarres R. Emicizumab Prophylaxis in Patients Who Have Hemophilia a without Inhibitors. New England Journal of Medicine 2018; 379: 811-22. https://www.nejm.org/doi/full/10.1056/NEJMoa1803550.
- Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, Santagostino E, Kruse-Jarres R, Negrier C, Kessler C, Valente N, Asikanius E, Levy GG, Windyga J, Shima M. Emicizumab Prophylaxis in Hemophilia a with Inhibitors. New England Journal of Medicine 2017; 377: 809-18. https://www.nejm.org/doi/full/10.1056/NEJMoa1703068.
- National Hemophilia Foundation. Masac Document 258 - Recommendation on the Use and Management of Emicizumab-Kxwh (Hemlibra®) for Hemophilia a with and without Inhibitors. National Hemophilia Foundation, 2020. https://www.hemophilia.org/healthcare-professionals/guidelines-on-care/masac-documents/masac-document-258-recommendation-on-the-use-and-management-of-emicizumab-kxwh-hemlibrar-for-hemophilia-a-with-and-without-inhibitors.
- Teitel J, Carcao M, Lillicrap D, Rivard GE, St-Louis J, Walker I, For the Inhibitor Subcommittee of the Association of Hemophilia Clinic Directors of Canada. A Guide to the Management of Patients with Inhibitors to Factor VIII and Factor IX. Association of Hemophilia Clinic Directors of Canada, 2010: p. 1-49. inhibitorguide2010.pdf (ahcdc.ca).
- Ljung RCR. How I Manage Patients with Inherited Haemophilia a and B and Factor Inhibitors. Br J Haematol 2018; 180: 501-10. https://www.ncbi.nlm.nih.gov/pubmed/29270992.
- Eckhardt CL, van Velzen AS, Peters M, Astermark J, Brons PP, Castaman G, Cnossen MH, Dors N, Escuriola-Ettingshausen C, Hamulyak K, Hart DP, Hay CRM, Haya S, van Heerde WL, Hermans C, Holmström M, Jimenez-Yuste V, Keenan RD, Klamroth R, Laros-van Gorkom BAP, Leebeek FWG, Liesner R, Mäkipernaa A, Male C, Mauser-Bunschoten E, Mazzucconi mg, McRae S, Meijer K, Mitchell M, Morfini M, Nijziel M, Oldenburg J, Peerlinck K, Petrini P, Platokouki H, Reitter-Pfoertner SE, Santagostino E, Schinco P, Smiers FJ, Siegmund B, Tagliaferri A, Yee TT, Kamphuisen PW, van der Bom JG, Fijnvandraat K, Group ftIS. Factor VIII Gene (F8) Mutation and Risk of Inhibitor Development in Nonsevere Hemophilia A. Blood 2013; 122: 1954-62. https://doi.org/10.1182/blood-2013-02-483263.
- Martinowitz U, Livnat T, Zivelin A, Kenet G. Concomitant Infusion of Low Doses of Rfviia and Feiba in Haemophilia Patients with Inhibitors. Haemophilia 2009; 15: 904-10. http://www.ncbi.nlm.nih.gov/pubmed/19473416.
- Croteau SE, Abajas YL, Wolberg AS, Nielsen BI, Marx GR, Baird CW, Neufeld EJ, Monahan PE. Recombinant Porcine Factor VIII for High-Risk Surgery in Paediatric Congenital Haemophilia a with High-Titre Inhibitor. Haemophilia 2017; 23: e93-e8. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.13157.
- Poon M-C, Goodyear MD, Rydz N, Lee A. Surgery in Mild Haemophilia a Patients with a History of Inhibitor Antibodies against Factor VIII: Individualized Management. Haemophilia 2021; 27: e768-e71. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.14415.
- Oldenburg J, Mahlangu JN, Kim B, Schmitt C, Callaghan MU, Young G, Santagostino E, Kruse-Jarres R, Negrier C, Kessler C, Valente N, Asikanius E, Levy GG, Windyga J, Shima M. Emicizumab Prophylaxis in Hemophilia a with Inhibitors. N Engl J Med 2017; 377: 809-18. https://www.ncbi.nlm.nih.gov/pubmed/28691557.
- Batsuli G, Zimowski KL, Tickle K, Meeks SL, Sidonio Jr RF. Immune Tolerance Induction in Paediatric Patients with Haemophilia a and Inhibitors Receiving Emicizumab Prophylaxis. Haemophilia 2019; 25: 789-96. https://onlinelibrary.wiley.com/doi/abs/10.1111/hae.13819.
- Tiede A, Collins P, Knoebl P, Teitel J, Kessler C, Shima M, Di Minno G, d’Oiron R, Salaj P, Jiménez-Yuste V, Huth-Kühne A, Giangrande P. International Recommendations on the Diagnosis and Treatment of Acquired Hemophilia A. Haematologica 2020; 105: 1791-801. https://haematologica.org/article/view/9931.
- Franchini M, Lippi G. Acquired Factor VIII Inhibitors. Blood 2008; 112: 250-5. https://doi.org/10.1182/blood-2008-03-143586.
- Kruse-Jarres R, St-Louis J, Greist A, Shapiro A, Smith H, Chowdary P, Drebes A, Gomperts E, Bourgeois C, Mo M, Novack A, Farin H, Ewenstein B. Efficacy and Safety of OBI-1, an Antihaemophilic Factor VIII (Recombinant), Porcine Sequence, in Subjects with Acquired Haemophilia A. Haemophilia 2015; 21: 162-70. https://www.ncbi.nlm.nih.gov/pubmed/25623166.
- Martin K, Kasthuri R, Mooberry MJ, Chen SL, Key NS, Ma AD. Lower Doses of Recombinant Porcine Factor VIII Maintain Excellent Haemostatic Efficacy. Haemophilia 2016; 22: e549-e51. https://www.ncbi.nlm.nih.gov/pubmed/27704655.
- Young G, Mahlangu JN. Extended Half-Life Clotting Factor Concentrates: Results from Published Clinical Trials. Haemophilia 2016; 22 Suppl 5: 25-30. https://pubmed.ncbi.nlm.nih.gov/27405672/.
- Negrier C, Knobe K, Tiede A, Giangrande P, Moss J. Enhanced Pharmacokinetic Properties of a Glycopegylated Recombinant Factor IX: A First Human Dose Trial in Patients with Hemophilia B. Blood 2011; 118: 2695-701. https://pubmed.ncbi.nlm.nih.gov/21555744/.
- Podust VN, Balan S, Sim BC, Coyle MP, Ernst U, Peters RT, Schellenberger V. Extension of in Vivo Half-Life of Biologically Active Molecules by XTEN Protein Polymers. J Control Release 2016; 240: 52-66. https://www.ncbi.nlm.nih.gov/pubmed/26497931.
- Pestel S, Beltz H-W, Claar P, Lind H, Mischnik M, Raquet E, Andrews A, Simmonds J, Tomasetig V, Dower SK, Tjärnlund-Wolf A, Schulte S, Schmidt PM, Weimer T. FVIII Half-Life Extension by Coadministration of a D′D3 Albumin Fusion Protein in Mice, Rabbits, Rats, and Monkeys. Blood Adv 2020; 4: 1870-80. https://doi.org/10.1182/bloodadvances.2019000999.
- Konkle BA, Shapiro AD, Quon DV, Staber JM, Kulkarni R, Ragni MV, Chhabra ES, Poloskey S, Rice K, Katragadda S, Rudin D, Fruebis J, Benson CC. Bivv001 Fusion Protein as Factor VIII Replacement Therapy for Hemophilia A. New England Journal of Medicine 2020; 383: 1018-27. https://www.nejm.org/doi/full/10.1056/NEJMoa2002699.
- Lauritzen B, Bjelke M, Björkdahl O, Bloem E, Keane K, Kjalke M, Rossen M, Lippert SL, Weldingh KN, Skydsgaard M, Kjellev S. A Novel Next-Generation FVIIIa Mimetic, Mim8, Has a Favorable Safety Profile and Displays Potent Pharmacodynamic Effects: Results from Safety Studies in Cynomolgus Monkeys. Journal of Thrombosis and Haemostasis 2022; 20: 1312-24. https://onlinelibrary.wiley.com/doi/abs/10.1111/jth.15682.
- Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T, Bevan D, Austin S, Hay CR, Hegemann I, Kazmi R, Chowdary P, Gercheva-Kyuchukova L, Mamonov V, Timofeeva M, Soh CH, Garg P, Vaishnaw A, Akinc A, Sorensen B, Ragni MV. Targeting of Antithrombin in Hemophilia A or B with RNAi Therapy. N Engl J Med 2017; 377: 819-28. https://www.ncbi.nlm.nih.gov/pubmed/28691885.
- Pasi KJ LT, Georgiev P, Mamonov V, Ragni MV, Yu Q, Andersson S, Johnson J, Ali S, Mei B,. Fitusiran, an Rnai Therapeutic Targeting Antithrombin to Restore Hemostatic Balance in Hemophilia: Interim Analysis from Open-Label Extension Study. In Research and Practice in Thrombosis and Haemostasis, Vol 3. Published by International Society for Thrombosis and Hemostasis, 2019.
- Chowdary P, Lethagen S, Friedrich U, Brand B, Hay C, Abdul Karim F, Klamroth R, Knoebl P, Laffan M, Mahlangu J, Miesbach W, Dalsgaard Nielsen J, Martin-Salces M, Angchaisuksiri P. Safety and Pharmacokinetics of Anti-Tfpi Antibody (Concizumab) in Healthy Volunteers and Patients with Hemophilia: A Randomized First Human Dose Trial. J Thromb Haemost 2015; 13: 743-54. https://www.ncbi.nlm.nih.gov/pubmed/25641556.
- Shapiro AD, Angchaisuksiri P, Astermark J, Benson G, Castaman G, Chowdary P, Eichler H, Jiménez-Yuste V, Kavakli K, Matsushita T, Poulsen LH, Wheeler AP, Young G, Zupancic-Salek S, Oldenburg J. Subcutaneous Concizumab Prophylaxis in Hemophilia a and Hemophilia A/B with Inhibitors: Phase 2 Trial Results. Blood 2019; 134: 1973-82. https://doi.org/10.1182/blood.2019001542.
- Mahlangu J LJ, Morales JC, Malan DR, Zupancic-Salek S, Wang M, Boggio LN, Hegemann I, Mital A, Cardinal M, Zhu T, Sun P, Arkin S,. A Phase 1b/2 Study of the Safety, Tolerability, Pharmacokinetics, Pharmacodynamics, and Efficacy of Pf-06741086, an Anti-TFPI Monoclonal Antibody, in Patients with Severe Hemophilia a or B. In Research and Practice in Thrombosis and Haemostasis, Vol3. Published by International Society for Thrombosis and Hemostasis, 2019.
- Zhao X-Y, Yegneswaran S, Bauzon M, Sim D, Patel C, Schneider D, McLean K, Zhu Y, Jiang X, Evans V, Gu J-M, Ivens I, Xu J, Bringmann PW, Kauser K, Esmon C. Targeted Inhibition of Activated Protein C Anticoagulant Activity by Monoclonal Antibody Hapc1573 for Treatment of Hemophilia. Blood 2016; 128: 80-. https://doi.org/10.1182/blood.V128.22.80.80.
- Polderdijk SGI, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and Characterization of an APC-Specific Serpin for the Treatment of Hemophilia. Blood 2017; 129: 105-13. https://doi.org/10.1182/blood-2016-05-718635.
- Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, Della Peruta M, Lheriteau E, Patel N, Raj D, Riddell A, Pie J, Rangarajan S, Bevan D, Recht M, Shen YM, Halka kg, Basner-Tschakarjan E, Mingozzi F, High KA, Allay J, Kay MA, Ng CY, Zhou J, Cancio M, Morton CL, Gray JT, Srivastava D, Nienhuis AW, Davidoff AM. Long-Term Safety and Efficacy of Factor IX Gene Therapy in Hemophilia B. N Engl J Med 2014; 371: 1994-2004. https://www.ncbi.nlm.nih.gov/pubmed/25409372.
- George LA, Sullivan SK, Giermasz A, Rasko JEJ, Samelson-Jones BJ, Ducore J, Cuker A, Sullivan LM, Majumdar S, Teitel J, McGuinn CE, Ragni MV, Luk AY, Hui D, Wright JF, Chen Y, Liu Y, Wachtel K, Winters A, Tiefenbacher S, Arruda VR, van der Loo JCM, Zelenaia O, Takefman D, Carr ME, Couto LB, Anguela XM, High KA. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N Engl J Med 2017; 377: 2215-27. https://www.ncbi.nlm.nih.gov/pubmed/29211678.
- Rangarajan S, Walsh L, Lester W, Perry D, Madan B, Laffan M, Yu H, Vettermann C, Pierce GF, Wong WY, Pasi KJ. AAV5-Factor VIII Gene Transfer in Severe Hemophilia A. N Engl J Med 2017; 377: 2519-30. https://www.ncbi.nlm.nih.gov/pubmed/29224506.
- Pasi KJ, Rangarajan S, Mitchell N, Lester W, Symington E, Madan B, Laffan M, Russell CB, Li M, Pierce GF, Wong WY. Multiyear Follow-up of AAV5-hFVIII-SQ Gene Therapy for Hemophilia A. New England Journal of Medicine 2020; 382: 29-40. https://www.nejm.org/doi/full/10.1056/NEJMoa1908490.
- Ozelo MC, Mahlangu J, Pasi KJ, Giermasz A, Leavitt AD, Laffan M, Symington E, Quon DV, Wang J-D, Peerlinck K, Pipe SW, Madan B, Key NS, Pierce GF, O’Mahony B, Kaczmarek R, Henshaw J, Lawal A, Jayaram K, Huang M, Yang X, Wong WY, Kim B. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. New England Journal of Medicine 2022; 386: 1013-25. https://www.nejm.org/doi/full/10.1056/NEJMoa2113708.
- Batty P, Lillicrap D. Hemophilia Gene Therapy: Approaching the First Licensed Product. HemaSphere 2021; 5: e540-e. https://pubmed.ncbi.nlm.nih.gov/33604517.
- Leebeek FW, Eikenboom JC. Von Willebrand's Disease. N Engl J Med 2016; 375: 2067-80. https://www.ncbi.nlm.nih.gov/pubmed/27959741.
- James PD, Connell NT, Ameer B, Di Paola J, Eikenboom J, Giraud N, Haberichter S, Jacobs-Pratt V, Konkle B, McLintock C, McRae S, R. Montgomery R, O’Donnell JS, Scappe N, Sidonio R, Jr, Flood VH, Husainat N, Kalot MA, Mustafa RA. Ash ISTH NHF WFH 2021 Guidelines on the Diagnosis of Von Willebrand Disease. Blood Adv 2021; 5: 280-300. https://doi.org/10.1182/bloodadvances.2020003265.
- Connell NT, Flood VH, Brignardello-Petersen R, Abdul-Kadir R, Arapshian A, Couper S, Grow JM, Kouides P, Laffan M, Lavin M, Leebeek FWG, O’Brien SH, Ozelo MC, Tosetto A, Weyand AC, James PD, Kalot MA, Husainat N, Mustafa RA. ASH ISTH NHF WFH 2021 Guidelines on the Management of Von Willebrand Disease. Blood Adv 2021; 5: 301-25. https://doi.org/10.1182/bloodadvances.2020003264.
- Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, Mainwaring J, Mathias M, O'Connell N, Committee B. Guideline for the Diagnosis and Management of the Rare Coagulation Disorders: A United Kingdom Haemophilia Centre Doctors' Organization Guideline on Behalf of the British Committee for Standards in Haematology. Br J Haematol 2014; 167: 304-26. https://www.ncbi.nlm.nih.gov/pubmed/25100430.
- Peyvandi F, Menegatti M. Treatment of Rare Factor Deficiencies in 2016. Hematology Am Soc Hematol Educ Program 2016; 2016: 663-9. https://www.ncbi.nlm.nih.gov/pubmed/27913544.
- Bolton-Maggs PH, Chalmers EA, Collins PW, Harrison P, Kitchen S, Liesner RJ, Minford A, Mumford AD, Parapia LA, Perry DJ, Watson SP, Wilde JT, Williams MD, UKHCDO. A Review of Inherited Platelet Disorders with Guidelines for Their Management on Behalf of the UKHCDO. Br J Haematol 2006; 135: 603-33. https://www.ncbi.nlm.nih.gov/pubmed/17107346.
- Israels SJ, Kahr WH, Blanchette VS, Luban NL, Rivard GE, Rand ml. Platelet Disorders in Children: A Diagnostic Approach. Pediatr Blood Cancer 2011; 56: 975-83. http://www.ncbi.nlm.nih.gov/pubmed/21294245.
- Lee A, Poon M-C. Inherited Platelet Functional Disorders: General Principles and Practical Aspects of Management. Transfus Apher Sci 2018.
- Rand ml, Reddy EC, Israels SJ. Laboratory Diagnosis of Inherited Platelet Function Disorders. Transfusion and Apheresis Science 2018; 57: 485-93. https://doi.org/10.1016/j.transci.2018.07.009.
- The Association of Hemophilia Clinic Directors of Canada. Diagnostic Criteria for Inherited Platelet Function Disorders for the Canadian Rare Inherited Bleeding Disorders Registry (RIBDR). 2013. https://www.ahcdc.ca/storage/files/diagnostic-criteria-2013.pdf.
- The Association of Hemophilia Clinic Directors of Canada. Algorithm for Analysis of Patients with Suspected Platelet Dysfunction2013. https://www.ahcdc.ca/storage/files/diagnostic-algorithm-2013.pdf (Last accessed 2018 August 10).
- Poon M-C, Di Minno G, d’Oiron R, Zotz R. New Insights into the Treatment of Glanzmann Thrombasthenia. Transfus Med Rev 2016; 30: 92-9. https://www.sciencedirect.com/science/article/pii/S088779631600002X.
- Tarawah A, Owaidah T, Al-Mulla N, Khanani M, Elhazmi J, Albagshi M, Wali Y, AlMohareb S, Almomen A. Management of Glanzmann's Thrombasthenia - Guidelines Based on an Expert Panel Consensus from Gulf Cooperation Council Countries. Journal of Applied Hematology 2019; 10: 1-9. https://www.jahjournal.org/article.asp?issn=1658-5127;year=2019;volume=10;issue=1;spage=1;epage=9;aulast=Tarawah.
- Di Minno G, Coppola A, Di Minno MND, Poon M-C. Glanzmann’s Thrombasthenia (Defective Platelet Integrin αIIb-β3): Proposals for Management between Evidence and Open Issues. Thromb Haemost 2009; 102: 1157-64.
- Poon M-C. The Use of Recombinant Activated Factor Vii in Patients with Glanzmann's Thrombasthenia. Thromb Haemost 2021; 121: 332-40.
- Poon M-C, d'Oiron R. Alloimmunization in Congenital Deficiencies of Platelet Surface Glycoproteins: Focus on Glanzmann's Thrombasthenia and Bernard–Soulier's Syndrome. Semin Thromb Hemost 2018; 44: 604-14. https://pubmed.ncbi.nlm.nih.gov/29879742/.
- Poon M-C, D'Oiron R, Von Depka M, Khair K, Négrier C, Karafoulidou A, Huth-Kuehne A, Morfini M, for members of the International Data Collection on Recombinant Factor VIIa and Congenital Platelet Disorders Study Group. Prophylactic and Therapeutic Recombinant Factor Viia Administration to Patients with Glanzmann's Thrombasthenia: Results of an International Survey. Journal of Thrombosis and Haemostasis 2004; 2: 1096-103. https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1538-7836.2004.00767.x.
- National Advisory Committee on Blood and Blood Products. Recommendations for Use of Prothrombin Complex Concentrates in Canada Available From: Http://Www.Nacblood.Ca/Resources/Guidelines/Pcc.Html. 2014.
- Tomaselli GF, Mahaffey KW, Cuker A, Dobesh PP, Doherty JU, Eikelboom JW, Florido R, Hucker W, Mehran R, Messe SR, Pollack CV, Jr., Rodriguez F, Sarode R, Siegal D, Wiggins BS. 2017 ACC Expert Consensus Decision Pathway on Management of Bleeding in Patients on Oral Anticoagulants: A Report of the American College of Cardiology Task Force on Expert Consensus Decision Pathways. J Am Coll Cardiol 2017; 70: 3042-67. https://www.ncbi.nlm.nih.gov/pubmed/29203195.
- Tornkvist M, Smith JG, Labaf A. Current Evidence of Oral Anticoagulant Reversal: A Systematic Review. Thromb Res 2018; 162: 22-31. https://www.ncbi.nlm.nih.gov/pubmed/29258056.
- Shih A, Nahirniak S, Pavenski K, Prokopchuk-Gauk O, Shabani-Rad M-T, Webert K. Recommendations for Use of Prothrombin Complex Concentrates in Canada. National Advisory Committee on Blood and Blood Products, 2022. https://nacblood.ca/en/resource/recommendations-use-prothrombin-complex-concentrates-canada.
- Kaatz S, Mahan CE, Nakhle A, Gunasekaran K, Ali M, Lavender R, Paje DG. Management of Elective Surgery and Emergent Bleeding with Direct Oral Anticoagulants. Curr Cardiol Rep 2017; 19: 124. https://www.ncbi.nlm.nih.gov/pubmed/29064044.
- Thrombosis Canada. Clinical Guides,2021. https://thrombosiscanada.ca/clinicalguides/ (Last accessed June 13, 2022.
- Schulman S, Ritchie B, Nahirniak S, Gross PL, Carrier M, Majeed A, Hwang HG, Zondag M, Study Investigators. Reversal of Dabigatran-Associated Major Bleeding with Activated Prothrombin Concentrate: A Prospective Cohort Study. Thromb Res 2017; 152: 44-8. https://www.ncbi.nlm.nih.gov/pubmed/28222322.
- Schulman S, Gross PL, Ritchie B, Nahirniak S, Lin Y, Lieberman L, Carrier M, Crowther MA, Ghosh I, Lazo-Langner A, Zondag M, Study I. Prothrombin Complex Concentrate for Major Bleeding on Factor Xa Inhibitors: A Prospective Cohort Study. Thromb Haemost 2018. https://www.ncbi.nlm.nih.gov/pubmed/29564837.
- Levi M, Scully M. How I Treat Disseminated Intravascular Coagulation. Blood 2018; 131: 845-54. https://www.ncbi.nlm.nih.gov/pubmed/29255070.
- Taylor FB, Jr., Toh CH, Hoots WK, Wada H, Levi M. Towards Definition, Clinical and Laboratory Criteria, and a Scoring System for Disseminated Intravascular Coagulation. Thromb Haemost 2001; 86: 1327-30. https://pubmed.ncbi.nlm.nih.gov/11816725/.
- Bakhtiari K, Meijers JC, de Jonge E, Levi M. Prospective Validation of the International Society of Thrombosis and Haemostasis Scoring System for Disseminated Intravascular Coagulation. Crit Care Med 2004; 32: 2416-21.
- Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ, Jr., Recombinant human protein CWEiSSsg. Efficacy and Safety of Recombinant Human Activated Protein C for Severe Sepsis. N Engl J Med 2001; 344: 699-709. https://www.ncbi.nlm.nih.gov/pubmed/11236773.
- Ranieri VM, Thompson BT, Barie PS, Dhainaut JF, Douglas IS, Finfer S, Gardlund B, Marshall JC, Rhodes A, Artigas A, Payen D, Tenhunen J, Al-Khalidi HR, Thompson V, Janes J, Macias WL, Vangerow B, Williams MD, PROWESS-SHOCK Study Group. Drotrecogin Alfa (Activated) in Adults with Septic Shock. N Engl J Med 2012; 366: 2055-64. https://www.ncbi.nlm.nih.gov/pubmed/22616830.
- Warren BL, Eid A, Singer P, Pillay SS, Carl P, Novak I, Chalupa P, Atherstone A, Penzes I, Kubler A, Knaub S, Keinecke HO, Heinrichs H, Schindel F, Juers M, Bone RC, Opal SM, KyberSept Trial Study Group. Caring for the Critically Ill Patient. High-Dose Antithrombin III in Severe Sepsis: A Randomized Controlled Trial. JAMA 2001; 286: 1869-78. https://www.ncbi.nlm.nih.gov/pubmed/11597289.
- Hayakawa M, Kudo D, Saito S, Uchino S, Yamakawa K, Iizuka Y, Sanui M, Takimoto K, Mayumi T, Ono K, Azuhata T, Ito F, Yoshihiro S, Hayakawa K, Nakashima T, Ogura T, Noda E, Nakamura Y, Sekine R, Yoshikawa Y, Sekino M, Ueno K, Okuda Y, Watanabe M, Tampo A, Saito N, Kitai Y, Takahashi H, Kobayashi I, Kondo Y, Matsunaga W, Nachi S, Miike T, Takahashi H, Takauji S, Umakoshi K, Todaka T, Kodaira H, Andoh K, Kasai T, Iwashita Y, Arai H, Murata M, Yamane M, Shiga K, Hori N. Antithrombin Supplementation and Mortality in Sepsis-Induced Disseminated Intravascular Coagulation: A Multicenter Retrospective Observational Study. Shock 2016; 46: 623-31. https://www.ncbi.nlm.nih.gov/pubmed/27548460.
- Hayakawa M, Yamakawa K, Saito S, Uchino S, Kudo D, Iizuka Y, Sanui M, Takimoto K, Mayumi T, Ono K, Japan Septic Disseminated Intravascular Coagulation study g. Recombinant Human Soluble Thrombomodulin and Mortality in Sepsis-Induced Disseminated Intravascular Coagulation. A Multicentre Retrospective Study. Thromb Haemost 2016; 115: 1157-66. https://www.ncbi.nlm.nih.gov/pubmed/26939575.
- Betschel S, Badiou J, Binkley K, Borici-Mazi R, Hébert J, Kanani A, Keith P, Lacuesta G, Waserman S, Yang B, Aygören-Pürsün E, Bernstein J, Bork K, Caballero T, Cicardi M, Craig T, Farkas H, Grumach A, Katelaris C, Longhurst H, Riedl M, Zuraw B, Berger M, Boursiquot J-N, Boysen H, Castaldo A, Chapdelaine H, Connors L, Fu L, Goodyear D, Haynes A, Kamra P, Kim H, Lang-Robertson K, Leith E, McCusker C, Moote B, O’Keefe A, Othman I, Poon M-C, Ritchie B, St-Pierre C, Stark D, Tsai E. The International/Canadian Hereditary Angioedema Guideline. Allergy, Asthma & Clinical Immunology 2019; 15: 72. https://doi.org/10.1186/s13223-019-0376-8.
- Rosen FS, Pensky J, Donaldson V, Charache P. Hereditary Angioneurotic Edema: Two Genetic Variants. Science 1965; 148: 957-8. https://www.ncbi.nlm.nih.gov/pubmed/14277836.
- Bouillet L, Boccon-Gibod I, Ponard D, Drouet C, Cesbron JY, Dumestre-Perard C, Monnier N, Lunardi J, Massot C, Gompel A. Bradykinin Receptor 2 Antagonist (Icatibant) for Hereditary Angioedema Type III Attacks. Ann Allergy Asthma Immunol 2009; 103: 448. https://www.ncbi.nlm.nih.gov/pubmed/19927548.
- Agostoni A, Cicardi M. Hereditary and Acquired C1-Inhibitor Deficiency: Biological and Clinical Characteristics in 235 Patients. Medicine (Baltimore) 1992; 71: 206-15. https://www.ncbi.nlm.nih.gov/pubmed/1518394.