Développement professionnel / Transfusion
Apprendre. Partager. Progresser
Jumelage de patients atteints d’anémie falciforme selon le phénotype et période de conservation du sang : examen et recommandations aux fins transfusionnelles
Il se peut que le langage utilisé dans cet article pour décrire la race et l’ethnicité ne reflètent pas les bonnes pratiques en matière de terminologie inclusive et respectueuse dans la recherche biomédicale.1-3 Dans le cadre de nos efforts constants pour actualiser le contenu destiné à la formation professionnelle, la terminologie utilisée dans cet article sera révisée pour s’aligner sur les meilleures pratiques de la communauté. Si vous avez des questions ou des commentaires, veuillez remplir ce formulaire.
Bibliographie
- Chew, M., Samuel, D., Mullan, Z., & Kleinert, S. (2024). The Lancet Group's new guidance to authors on reporting race and ethnicity. Lancet, 403(10442), 2360-2361. https://doi.org/10.1016/s0140-6736(24)01081-x
- Stanbrook, M. B., & Salami, B. (2023). CMAJ’s new guidance on the reporting of race and ethnicity in research articles. Canadian Medical Association Journal, 195(6), E236-E238. https://doi.org/10.1503/cmaj.230144
- National Academies of Sciences, E., & Medicine. (2025). Rethinking Race and Ethnicity in Biomedical Research. The National Academies Press. https://doi.org/doi:10.17226/27913
Auteurs : Pierre-Aurèle Morin, MD; Robert Skeate, MD, MSc; et Gwen Clarke, MD, FRCPC
Mise en ligne : avril 2020
Points essentiels
- Fréquente chez les patients atteints d’anémie falciforme ou drépanocytose, l’allo‑immunisation complique parfois la thérapie transfusionnelle.
- Afin de réduire les risques d’allo-immunisation, on recommande de déterminer le phénotype des patients et de les jumeler à un donneur compatible de façon préventive.
- Chez les personnes drépanocytaires, les exigences transfusionnelles et la présence ou non d’anticorps anti-érythrocytaires régissent l’ampleur du processus de détermination du phénotype et la pertinence du génotypage.
- Il existe une quantité restreinte de données probantes sur les conséquences de la période de conservation du sang au sein de cette population. Toutefois, l’approvisionnement en unités fraîches apporte son lot de difficultés considérables et pourrait nuire à la disponibilité d’unités à phénotype optimal.
Thérapie transfusionnelle chez les patients atteints d’anémie falciforme
La transfusion de globules rouges demeure incontournable pour la prise en charge des patients drépanocytaires. Elle est indiquée dans les cas de crise aplasique aiguë, de séquestration splénique ou hépatique aiguë et d’anémie symptomatique dans le traitement et la prévention des accidents cérébraux vasculaires (ACV) et du syndrome thoracique aigu, ainsi que dans la préparation aux chirurgies importantes. 1, 2 Si la plupart des indices de son efficacité sont de piètre qualité, des essais cliniques randomisés montrent que la transfusion préventive de globules rouges réduit la fréquence des ACV chez les patients pédiatriques drépanocytaires à risque.3,4
Ce type de thérapie se fait par transfusion simple ou par exsanguino-transfusion. Elle s’impose généralement chez les patients qui souffrent de complications aiguës graves, comme un ACV ou un syndrome thoracique aigu, et s’inscrit également dans le traitement préventif de certains patients à risque élevé. L’exsanguino-transfusion de globules rouges offre divers avantages, notamment l’augmentation minimale du taux de fer net, l’atteinte rapide et efficace des taux d’hémoglobine S cibles (~30 %) et la possibilité d’obtenir un hématocrite optimisant la viscosité sanguine. 5, 6 Parmi les désavantages, notons un nombre élevé de transfusions de globules rouges nécessaires, une exposition accrue à plusieurs donneurs, une augmentation des coûts et un besoin en équipement spécialisé et en expertise.7 L’exposition du transfusé à des donneurs multiples ne semble pas augmenter le risque d’allo-immunisation. 8 De plus, le service de transfusion aura comme défi de trouver de nombreuses unités dépourvues d’antigènes dans une même journée. Cela est d’autant plus vrai en ce qui a trait aux demandes d’unités à courte période de conservation.
Taux d’allo-immunisation chez les patients drépanocytaires
Il n’est pas rare que les patients drépanocytaires deviennent immunisés contre les antigènes érythrocytaires.9 Après avoir examiné 12 études, Garratty a conclu que l’allo-immunisation se manifesterait entre 8 % et 35 % des patients drépanocytaires transfusés, avec une médiane de 25 %.10 L’allo-immunisation nuit aux soins puisqu’elle prolonge le délai de transfusion et entraîne parfois des complications importantes, comme une réaction transfusionnelle hémolytique retardée.11 Garratty fait remarquer que les patients drépanocytaires peuvent subir de graves réactions et connaître d’autres complications associées, comme des crises de douleur.10 En de rares occasions, ils peuvent faire une hyperhémolyse à la suite d’une transfusion. L’hyperhémolyse se caractérise par une hémolyse rapide des globules rouges du donneur et du receveur et par l’exacerbation de l’hémolyse au cours des transfusions subséquentes. Étant donné que ce type de complication n’est pas toujours lié à la présence d’anticorps anti‑érythrocytaires, d’autres études seront nécessaires pour confirmer le lien entre l’hyperhémolyse et l’allo-immunisation.12
Chez les patients drépanocytaires, les antigènes les plus souvent associés à l’allo-immunisation comprennent les antigènes RhC, RhE et Kell, entre autres.13 Tiré d’un article bien connu de Vichinsky et coll., paru dans le New England Journal of Medicine en 1990, le tableau 1 montre la fréquence de différents allo-anticorps chez 32 patients drépanocytaires transfusés sur un total de 107 patients, et 17 d’entre eux présentaient plus d’un anticorps.14
Tableau 1 : Distribution de 68 alloanticorps anti-érythrocytaires chez 107 patients receveurs de transfusion pour anémie falciforme14
| Anticorps | N bre (%) |
|---|---|
| K | 18 (26) |
| E | 16 (24) |
| C | 11 (16) |
| Jkb | 7 (10) |
| Fya | 4 (6) |
| M | 3 (4) |
| Lea | 3 (4) |
| S | 2 (3) |
| Fyb | 2 (3) |
| e | 1 (2) |
| Jka | 1 (2) |
Parmi les facteurs contribuant aux taux élevés d’allo-immunisation, mentionnons que les phénotypes érythrocytaires des donneurs (prédominance de Blancs) ne correspondent pas à la population des patients drépanocytaires (non-Blancs, principalement de descendance africaine), d’où le taux élevé d’antigènes incompatibles entre donneur et receveur.14 Par exemple, Castro et ses collègues ont analysé les cas de sensibilisation qui auraient pu être évités dans un groupe de 137 patients drépanocytaires allo-immunisés si diverses stratégies préventives de jumelage phénotypique avaient été disponibles. L’analyse visait notamment à calculer la fréquence prévue des phénotypes requis pour jumeler les patients aux donneurs de sang blancs et afro-américains (colonnes de droite du tableau 2).15 Les colonnes « Donneurs blancs » et « Donneurs afro-américains » indiquent les différences de fréquence des phénotypes pertinents pour chacun des deux groupes et soulignent la façon dont le traitement de patients drépanocytaires au moyen de sang provenant de donneurs blancs pourrait accroître le risque d’allo-immunisation.
Tableau 2 : Projections relatives à la prévention de l’allo-immunisation chez les patients drépanocytaires ayant reçu des transfusions, en fonction de différents protocoles de jumelage phénotypique15
| Protocole de jumelage | n (%)* | Patients drépanocytaires chez qui un protocole aurait pu éviter la production d’alloanticorps, n (%)† | Phénotype | Exigences pour le jumelage de donneurs selon leur phénotype Fréquence des phénotypes ‡ chez |
|
|---|---|---|---|---|---|
| Donneurs blancs (%) | Donneurs afro-américains (%) | ||||
| ABO et D seulement | Aucun (étude en cours) | 249 (70,9) | ABO et D seulement | s. o. | s. o. |
| Protocole 1: D, C, c. E, e | 51 (37,2) | 289 (82,3) |
D+C-c+E-e+(R0)§ ou |
3,2 |
42,3 |
| D-C-c+E-e+(rr)§ | 15,0 | s. o. | |||
| Protocole 2: D, C, c, E, e, K | 73 (53,3) | 307 (87,5) | D-C-c+E-e+, K- | 13,6 | 41,2 |
| Protocole 3: D, C, c, E, e, K, S | 76 (55,5) | 310 (88,3) | D-C-c+E-e+, K-, S- | 6,1 | 28,4 |
| Protocole 4: D, C, c, E, e, K, S, Fy2 | 86 (62,8) | 320 (91,2) | D-C-c+E-e+, K-, S-, Fy(a-) | 2,1 | 14,6 |
|
Pourcentage des 137 patients drépanocytaires receveurs de transfusion ayant développé des alloanticorps. ‡Fréquence des phénotypes calculée selon les tableaux du manuel technique et fréquence des donneurs par thrombaphérèse non sélectionnés de type D-. †Nombre total de patients receveurs de transfusion (351) utilisé comme dénominateur. §La plupart des cliniques de transfusion donnent des unités de GR D-(rr) aux receveurs D+ en attente de GR C-E. |
|||||
Sans stratégie préventive de jumelage en fonction des antigènes, l’allo-immunisation touche environ 30 % des patients drépanocytaires. À l’aide de prévisions de la fréquence d’antigènes chez les donneurs et les receveurs, des protocoles rigoureux de jumelage de phénotypes entraîneront une réduction substantielle de l’allo-immunisation (voir tableau 2). Cependant, les avantages de l’ajout d’antigènes supplémentaires diminuent à mesure que les stratégies s’intensifient (p. ex., amélioration de 2,2 % entre les protocoles 3 et 4).
Jumelage d’antigènes visant à réduire l’allo-immunisation
Tahhan et ses collaborateurs ont comparé rétrospectivement les taux d’allo-immunisation de 40 patients ayant reçu des transfusions de sang dont les antigènes étaient compatibles avec les leurs (C, E, Kell, S, Fya, Fyb) et ceux de 46 patients ayant reçu des transfusions de sang dont les antigènes étaient compatibles ou non avec les leurs.16 Le taux d’allo-immunisation du premier groupe était nul, alors que celui du second atteignait 16 %.
Ameen et ses collègues ont effectué une analyse comparative semblable chez des patients koweïtiens, où le premier groupe a reçu du sang non jumelé (groupe 1, 110 patients), et où le second a reçu du sang dont les antigènes érythrocytaires C, c, E, e, et Kell étaient compatibles (groupe 2, 123 patients).17 Le taux d’allo-immunisation du premier groupe s’élevait à 65 %, et celui du second, à 23,6 %. On a observé ces taux élevés dans un groupe ethnique pourtant homogène. Les auteurs ont conclu à l’importance du jumelage des antigènes en cas de drépanocytose.
Vichinsky et son équipe a vérifié de façon officielle l’hypothèse voulant que le jumelage préventif des antigènes réduise la fréquence de l’allo-immunisation chez les patients drépanocytaires grâce à une analyse secondaire dans le cadre de l’étude STOP.18 Les 63 patients répartis aléatoirement dans le groupe transfusionnel ont reçu 1 830 transfusions de GR sur une période d’environ 21 mois. Ces unités étaient compatibles avec les antigènes C, E et Kell (en plus des antigènes de routine ABO et D). Seulement 29 unités transfusées n’avaient subi aucun jumelage autre que pour les antigènes ABO et D, ce qui a exposé 11 patients sur 63 (16 %) à des unités non jumelées. Ces patients ont développé de nouveaux anticorps (5 % d’anticorps chauds, 3 % d’anticorps de faible importance sur le plan clinique, 8 % de nouveaux allo-anticorps). Malgré les efforts pour fournir du sang compatible avec les antigènes C, E, et Kell, 4 des 5 patients ayant produit de nouveaux alloanticorps en avaient de type anti-E ou anti-Kell, dont 1 patient a fabriqué des anticorps anti-Fya et anti-S. Les chercheurs ont déterminé que le taux de formation de nouveaux anticorps dans le cadre de cette étude était de 0,5 % par unité transfusée, comparativement au taux de 3 % pour les patients ayant reçu des GR non jumelés, qu’ils ont calculé à partir de publications antérieures. Le taux de réaction transfusionnelle hémolytique a également substantiellement diminué. Ils ont conclu à la faisabilité et à l’utilité du jumelage préventif des antigènes C, E et Kell chez les patients drépanocytaires.
Un groupe de chercheurs a effectué une étude de suivi de deux ans auprès des participants à l’étude STOP.19 Les 78 patients ont bénéficié d’une thérapie transfusionnelle régulière après la fin de l’étude et ont reçu du sang aux antigènes compatibles. Les taux d’allo-immunisation sont demeurés faibles (taux de 0,5 % de formation de nouveaux anticorps par unité transfusée) tout au long de l’analyse supplémentaire de deux ans.
Lasalle-Williams et ses collègues ont présenté les résultats de 14 années de jumelage d’antigènes à leur établissement.20 De 1993 à 2006, ils ont transfusé des GR aux antigènes compatibles à 99 patients drépanocytaires (6 946 unités compatibles aux systèmes Rh (C, c, D, E, e), Kell (K, k), Duffy (Fya, Fyb), Kidd (Jka, Jkb), Lewis (Lea, Leb) et MNS (M, N, S, s)). Dans certains cas, on a privilégié une discordance entre les antigènes Lewis et MNS lorsqu’il était impossible d’obtenir une compatibilité exacte. Ils ont obtenu un taux d’allo-immunisation de 7 %. Dans leur rapport, ils ont présenté un tableau résumant une grande partie des études traitant des effets du jumelage d’antigènes sur les patients drépanocytaires (voir tableau 3). De façon générale, le sang non jumelé semble produire un taux d’allo-immunisation d’environ 30 %, contre un taux d’environ 10 % pour le sang jumelé.
Tableau 3 : Études évaluant le taux d’allo-immunisation et le jumelage d’antigènes de GR20
| Jumelage ABO et D seulement | ||
|---|---|---|
| Auteurs | Nbre de patients/de transfusions | % d’allo-immunisation/nbre d’alloanticorps par 100 unités transfusées |
|
Ambruso et al. |
85/1 941 |
34 %/3,4
|
| Rosse et al. | 1 044/----* | 18 %-31 % (27 % dans le groupe expérimental)/----- |
| Vichinsky et al. | 107/---- | 30%/----- |
| Aygun et al. |
140/3 239 (patients adultes et pédiatriques) |
37 %/2,8 |
| Castro et al. | 351/8 939 | 29 %-35 %/3,8 |
| Sakhalkar et al | 387/14 263 | 31 %/1,7 |
| Jumelage ABO, D, C, E, K | ||
| Auteurs | Nbre de patients/de transfusions | % d’allo-immunisation/taux d’alloanticorps par 100 unités transfusées |
| Vichinsky et al. |
Jumelage supplémentaire : C, E, K 61/1 830 |
8 %-11 %/0,5 |
| Sakhalkar et al. |
Jumelage supplémentaire : C, E, K 113/2 345 |
5 %/0,26 |
| Jumelage ABO, D, C, E, K et autres | ||
| Auteurs | Nbre de patients/de transfusions | % d’allo-immunisation/taux d’alloanticorps par 100 unités transfusées |
| Tahhan et al. |
Jumelage supplémentaire : C, E, K, S Fya , Fyb 40/----- |
0/------ |
| *Un trait indique des données manquantes | ||
Publié en 2019, un examen systématique de l’incidence du jumelage d’antigènes21 a recensé 19 études à ce sujet. Toutes les études contenaient des données d’observation et aucun essai clinique randomisé n’a été réalisé. En tout, 15 études évaluent le jumelage phénotypique sur l’allo-immunisation, l’auto-immunisation et les réactions aux transfusions; bien que les résultats soient de piètre qualité, leurs conclusions suggèrent que le jumelage sérologique d’antigènes de GR pourrait diminuer le risque d’allo-immunisation, mais le compte rendu des réactions aux transfusions manque de substance. Les données sont insuffisantes pour évaluer les répercussions financières de cette pratique sur le coût et la disponibilité des unités, ainsi que les effets du jumelage génotypique d’antigènes de GR. Les auteurs ont mis l’accent sur la nécessité de réaliser des essais cliniques prospectifs multicentriques pour déterminer les effets du jumelage et pour fournir des données probantes à l’appui d’une pratique clinique du jumelage.
Pratiques cliniques en matière de jumelage d’antigènes aux États-Unis
Depuis le début des années 2000, le jumelage phénotypique est devenu pratique courante aux États-Unis. Publié en 2016, un sondage22 a été réalisé dans 90 services de transfusion répartis dans diverses régions des États-Unis, dont 76 % étaient issus du milieu universitaire et 24 %, d’hôpitaux communautaires. Dans ces derniers, les activités de transfusion étaient grandement hétérogènes, étant donné que 52 % d’entre eux prodiguaient des soins transfusionnels à tout au plus 30 patients drépanocytaires. Parmi les services de transfusion, 90 % disposaient d’une politique relative au jumelage phénotypique; 74 % étaient compatibles à Cc, Ee et K; 13 % ont effectué un jumelage phénotypique exhaustif et 3 % ont adopté d’autres politiques.
Ces renseignements présentent un contraste flagrant aux données qui avaient été recueillies en 2003,23 où seuls 37 % des centres se servaient couramment du jumelage phénotypique dans les cas d’anémie falciforme.
Pratiques cliniques en matière de jumelage d’antigènes au Canada
Nous avons posé des questions informelles à certains experts en médecine transfusionnelle canadiens qui exercent leur profession au sein d’hôpitaux où l’on soigne régulièrement des patients drépanocytaires. À Toronto, le Hospital for Sick Children et les hôpitaux du University Health Network procèdent au phénotypage (et au génotypage si possible) des patients drépanocytaires relativement aux antigènes C, c, E, e, K, k, Fya, Fyb, Jka, Jkb, S et s. Si un patient ne présente pas d’allo-immunisation, ils effectuent le jumelage préventif pour les antigènes C, c, E, e, K et k. En présence d’anticorps anti-érythrocytaires, les unités transfusées doivent être compatibles avec les antigènes C, c, E, e, K, k, Fya, Fyb, Jka, Jkb, S et s (antigènes négatifs pour la cible des anticorps présents). Les experts d’Edmonton font le phénotypage pour les antigènes C, c, E, e, K, k, Fya, Fyb, Jka, Jkb, S, s, M, N, Lua et Lub. À l’instar de leurs homologues de Toronto, ils fournissent des unités compatibles avec les antigènes C, c, E, e, K et k aux patients non allo-immunisés et des unités à compatibilité accrue aux patients qui produisent des anticorps. À Montréal, selon l’expert sondé, les patients non allo-immunisés reçoivent des unités compatibles avec les antigènes C, E et K, les patients qui n’ont développé qu’un antigène obtiennent des unités exemptes de l’antigène cible, et les patients qui fabriquent un deuxième anticorps d’importance sur le plan clinique se voient attribuer des unités jumelées selon leur phénotype complet.
Ces protocoles sont fort semblables et sous-entendent que les experts canadiens sont profondément d’accord en ce qui concerne la meilleure manière de procéder à la transfusion de patients drépanocytaires.
Dans Consensus Statement on the Care of Patients with Sickle Cell Disease in Canada,24 il y a des recommandations qui vont dans le sens de cette manière de procéder. En voici quelques exemples :
- Déterminer le phénotype complet des GR dès la première visite, ainsi que le génotype si possible.
- Chez les patients qui n’ont pas développé d’alloanticorps, choisissez des unités de GR compatibles avec le Rh (D, C, c, E, e) et l’antigène Kell (K) du patient.
- Chez les patients qui ont des alloanticorps, sélectionnez des unités de GR compatibles avec les antigènes Rh (D, C, c, E, e), Kell (K), Kidd (Jka, Jkb), Duffy (Fya, Fyb) et S (S, s) du patient, ainsi qu’à tout autre antigène pour lequel le patient a déjà produit des anticorps.
Tout récemment, le groupe International Collaboration for Transfusion Medicine Guidelines (ICTMG), qui réunit des experts en transfusion, dont des Canadiens, a également publié des lignes directrices relatives aux spécifications quant aux GR dans les hémoglobinopathies.25 Leurs recommandations sont les mêmes que celles qui ont été énoncées ci-dessus.
Génotypage des patients drépanocytaires
Les études du génotypage portant sur l’anémie falciforme sont primordiales dans le dépistage des patients possédant des variants RhCE et RhD. Ces variants génétiques peuvent provoquer une allo-immunisation à des antigènes « partiels » perçus comme des autoanticorps par l’organisme et peuvent compliquer l’analyse des anticorps ou la sélection de GR pour transfusion. Des résultats de génotypage connus peuvent rendre possible l’identification de nouveaux anticorps et sont susceptibles d’influencer le choix de GR aux fins de transfusion.
Publiées en 2020, les nouvelles lignes directrices de l’American Society of Hematology (ASH) sur l’anémie falciforme26 indiquent que les patients drépanocytaires risquent de développer des alloanticorps contre les antigènes Rh en dépit d’une compatibilité avec les antigènes C et E ou C, c, E et e. Ce risque est attribuable à la prévalence accrue de variants Rh chez les patients drépanocytaires. Les anticorps contre les variants RhD et RhCE peuvent se manifester sous forme d’autoanticorps ou d’alloanticorps. Lorsque cela s’avère possible, le jumelage moléculaire est la solution idéale, mais il est souvent difficile à réaliser. Le génotypage des antigènes RhD et RhCE est disponible à la Société canadienne du sang et il est vivement recommandé de recourir à ce service pour les patients drépanocytaires. En présence de variants et après la détection des anticorps associés, il est possible de lancer une recherche de donneurs à haute compatibilité.
De même, l’identification de patients drépanocytaires qui présentent une mutation de type GATA1 affectant l’expression de l’antigène Fyb représente une avancée importante dans le domaine de la génétique. Par ailleurs, s’il est vrai que des sujets à phénotype dépourvu d’antigènes Fyb présentent une absence d’antigène Fyb sur leurs GR, la vaste majorité d’entre eux possèdent des antigènes Fyb s’étant exprimés sur d’autres types de cellule.27 Par conséquent, ils ne risquent pas de produire des anti-Fyb. En pratique, cela permet aux médecins spécialisés en transfusion de choisir des unités contenant des antigènes Fyb pour les patients drépanocytaires sans antigènes Fyb, ce qui accroît grandement la disponibilité d’unités de GR compatibles avec ces patients.28, 29
Réponse à la demande d’unités phénotypées de la Société canadienne du sang
Il manque malheureusement de données probantes en ce qui concerne la transfusion de patients drépanocytaires. Si le pays compte bon nombre de résidents originaires des Caraïbes et de l’Afrique de l’Ouest, on ignore toujours la prévalence réelle de la maladie au Canada.30 En 2018, les données recueillies par le Groupe canadien d’aphérèse révèlent que 210 patients drépanocytaires ont subi plus de 1 300 échanges de GR (correspondance personnelle, 4 juillet 2019). Nous ne connaissons ni le nombre total de cas ni le taux de soumission de demandes de phénotypage pour les patients drépanocytaires au Canada.
L’expérience du personnel de la Société canadienne du sang et l’expérience des médecins transfusionnels correspondent à celle des centres spécialisés dans le traitement de l’anémie falciforme aux États-Unis, qui rendent compte des difficultés qu’ils éprouvent à répondre à la demande croissante d’unités compatibles pour leurs patients.31-39
Mesures en réponse à la demande d’unités phénotypées au Canada
Le programme national de phénotypage des antigènes de GR fournit des unités de GR, assorties d’une description des antigènes qu’elle contient, aux patients ayant développé des anticorps et à ceux pour qui un jumelage préventif d’antigènes est nécessaire. En plus du groupage ABO et RhD ordinaire, le sang de tout donneur est analysé lors du premier et du deuxième don pour déterminer la nature de l’antigène K. Le dépistage des antigènes RhC, c, E, e, Fya, Fyb, Jka, Jkb, S et s s’effectue à l’aide d’une méthode algorithmique visant à maximiser le nombre de donneurs possédant un phénotype précis, sans antigène et compatible avec des anticorps très répandus. En général, ce sont les donneurs de groupe O, A et B qui sont visés par le phénotypage. Certes, deux dons sont nécessaires pour confirmer le typage, mais le phénotype s’imprime sur l’étiquette de produit dès qu’un typage sans un antigène donné est disponible.
Image
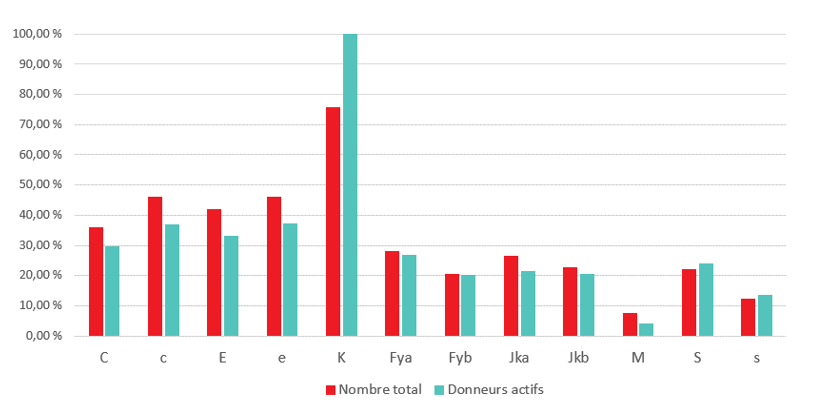
Diagramme 1 : Phénotypage à la Société canadienne du sang : Pourcentage du total des donneurs et de donneurs actifs phénotypés par antigène, janvier 2020.
Soucieuse de limiter l’incidence des demandes de phénotypage sur la disponibilité de GR RhD-, la Société canadienne du sang communique avec ses anciens donneurs R0R0 et R0R (D+, C- et E-) et cherche de nouveaux donneurs pour renflouer ses réserves d’unités C- et E-, souvent utilisées en cas de drépanocytose, et limiter le recours aux unités Rh- (rr; D-, C- et E -) afin de répondre à la demande. En outre, la Société procède au recrutement proactif et au phénotypage ciblé de donneurs issus de divers groupes ethniques pour déterminer la fréquence de différents phénotypes dans ces groupes, de sorte à répondre aux besoins de tous les Canadiens, quelles que soient leurs origines. Parallèlement, la Société est à même d’effectuer le génotypage de donneurs de certains phénotypes d’intérêt particulier (notamment S-s-) pour répondre à la demande croissante d’unités de sang rare (notamment U-) pour les patients drépanocytaires.
Période de conservation du sang aux fins d’exsanguino‑transfusion chez les patients drépanocytaires
Outre les demandes de sang à phénotype compatible, les demandes de sang frais sont monnaie courante en exsanguino-transfusion. Toutefois, on observe des variations considérables en pratique. En Ontario seulement, des grands centres ont commandé certaines unités fraîches de moins de 14 jours et d’autres dont la longueur de la période de conservation n’était pas limitée.
À ce jour, des études de cohorte d’envergure ont confirmé que dans la population générale, chez les patients ayant subi une chirurgie cardiaque, chez les personnes gravement malades et chez les prématurés, l’âge des unités de GR à transfuser n’a aucune influence sur les résultats cliniques.40-43 Néanmoins, certaines caractéristiques inhérentes à l’anémie falciforme sont susceptibles de rendre ces observations moins applicables. Également, plusieurs études ont révélé qu’avec le temps les GR subissent des modifications (aussi appelées « lésions d’entreposage »). Parmi ces modifications, mentionnons une réduction de la capacité de transport d’oxygène, une diminution de la déformabilité des membranes et une augmentation d’hémoglobine sans GR44 (associée à une baisse de la concentration en oxyde nitrique); ces modifications sont également attribuables à l’anémie falciforme en elle-même et on se demande si la maladie sous-jacente risque d’être d’exacerbée par la transfusion de GR à fraîcheur réduite. Cette inquiétude est encore plus présente lorsqu’il s’agit d’exsanguino-transfusion, durant laquelle un grand volume de sang est transfusé dans le but de remplacer les GR dysfonctionnels.
Quelques études se sont penchées sur les effets de l’âge du sang en cas d’anémie falciforme et la plupart n’abordent pas les patients qui ont recours à l’exsanguino-transfusion. Seule une étude de cohorte a traité partiellement de la question. En Ouganda, l’essai clinique TOTAL45 a débuté par la répartition aléatoire de 290 enfants en Ouganda (13 % d’entre eux étaient drépanocytaires) qui souffraient d’anémie grave accompagnée d’acidose lactique traitée à l’aide d’une transfusion de GR âgés soit de 25 à 35 jours, soit de 1 à 10 jours. On n’a constaté aucune différence entre les deux groupes ni à l’étape des résultats préliminaires (sur la proportion de patients présentant un niveau de lactate de moins de 3 mmol/l après 8 heures), ni à l’étape des résultats secondaires (résultats neurologiques et respiratoires, signes vitaux, fonction rénale, etc.).
Les études de cohorte ont obtenu des résultats variables, dont :
- Chez 131 patients drépanocytaires de 22 ans ou moins souffrant d’un syndrome thoracique aigu (234 épisodes) qui ont été traités à l’aide de transfusions régulières de GR, on n’a constaté aucun lien entre le temps de conservation des GR et la durée du séjour à l’hôpital et de l’oxygénothérapie nécessaire.46
- Chez 28 patients drépanocytaires adultes faisant l’objet d’un protocole permanent de transfusions régulières, la transfusion d’unités de ≥25 jours (par opposition à <25 jours) était associée à un risque accru d’infection, mais pas à des accès de douleur.47
- Chez 166 patients drépanocytaires transfusés d’après diverses indications (à l’exclusion de l’exsanguino-transfusion permanente), on a constaté une augmentation du risque d’allo-immunisation pour chaque période de 7 jours supplémentaire de conservation de GR (comparativement à un sujet non transfusé). Le ratio de risque de développer des anticorps anti-érythrocytaires 28 jours après la transfusion était de 3:5 pour les unités de 7 jours et de 9:8 pour les unités de 35 jours.48
Aucune de ces études n’aborde de patient qui reçoit des exsanguino-transfusions de façon permanente et la plupart des sujets n’ont reçu que des transfusions régulières. Soulignons qu’il y a un besoin flagrant d’essais cliniques de cohorte pour analyser le rôle que joue l’âge des unités de GR dans les résultats cliniques concernant les patients drépanocytaires transfusés.
Pratiques actuelles et recommandations officielles
Au mois de mai 2018, un sondage non officiel réalisé dans six grands hôpitaux de l’Ontario a révélé un écart marqué dans les pratiques actuelles, étant donné que les commandes d’unités concernaient des unités de <10 jours, de <14 jours, de <21 jours ou sans limite d’âge.
D’ailleurs, les lignes directrices divergent également quant aux recommandations sur l’âge des unités de GR transfusées et il n’y a aucune recommandation sur l’âge du sang transfusé dans la plupart d’entre elles. 24, 25, 49
Difficultés d’approvisionnement en sang frais
Il convient de rappeler que les unités de GR destinées aux patients drépanocytaires sont choisies en fonction de caractéristiques phénotypiques particulières, ce qui représente d’emblée un défi pour la Société canadienne du sang et pour d’autres fournisseurs de sang. En effet, les phénotypes érythrocytaires des personnes de descendance africaine diffèrent des phénotypes des Blancs, or la majorité du sang disponible au Canada est donné par des Blancs.
La situation devient d’autant plus difficile lorsque les commandes de GR précisent que la période de conservation du sang doit être inférieure à 14 jours. Près de 25 % du stock total de la plupart des centres de distribution de la Société est entreposé depuis plus de 14 jours, alors trouver des unités qui sont à la fois d’un âge d’entreposage défini et compatibles sur le plan phénotypique devient particulièrement ardu. Si une exsanguino-transfusion urgente est requise, l’approvisionnement en unités adéquates se complexifie encore plus. Dans ces cas-là, deux options sont possibles pour obtenir des unités répondant à toutes les exigences : recruter immédiatement des donneurs avec un phénotype compatible ou obtenir le transfert d’unités provenant d’autres centres au pays. Chacune mobilise des ressources humaines et financières considérables. De plus, elles créent un déséquilibre dans la distribution d’unités phénotypées en sol canadien, puisque les centres sans exigence concernant l’âge des unités s’en trouvent désavantagés, car ils seront confrontés à plus de demandes de transfert d’unités phénotypées vers d’autres centres, ce qui pourrait causer une pénurie d’unités de ce type.
Recommandations sur les spécifications relatives aux GR chez les patients drépanocytaires
En raison des difficultés liées aux besoins de transfusion chez les patients drépanocytaires au Canada, l’élaboration de lignes directrices générales s’avère utile pour l’optimisation de la gestion des demandes et l’établissement de priorités, surtout lorsque les réserves ne suffisent pas. D’autres facteurs doivent être pris en considération pour les patients qui présentent des caractéristiques complexes, comme ceux qui combinent un autoanticorps et un alloanticorps, et dans des situations particulières, comme la grossesse. Les recommandations seront appelées à changer avec l’obtention de nouvelles données cliniques et l’évolution des technologies, comme le génotypage des donneurs et des receveurs.
Recommandations concernant l’approvisionnement de sang à phénotype déterminé :
Patients n’ayant développé aucun anticorps
1. Déterminer le phénotype du patient.
2. Procéder au génotypage de chaque patient avant le début des transfusions afin de veiller à la compatibilité avec des antigènes partiels et au dépistage d’allèles rares.
3. Transfuser des GR ayant été soumis au jumelage préventif des antigènes C, c, E, e et K.
Patients ayant développé un ou plusieurs anticorps
1. Déterminer le phénotype du patient.
2. Envisager sérieusement de procéder au génotypage du patient s’il n’a pas déjà été réalisé.
3. Transfuser les globules rouges compatibles avec les antigènes importants sur le plan clinique pour lesquels des anticorps ont été identifiés. Puisque le système immunitaire de ces patients a déjà eu ce type de réaction, il est probable qu’ils développent des anticorps contre d’autres antigènes. Il serait donc judicieux d’effectuer le jumelage préventif des antigènes Fya, Fyb (s’ils sont de vrais négatifs), Jka, Jkb, S et s, en plus de C, c, E, e et K, si possible.
Ces recommandations se conforment aux lignes directrices de l’Association canadienne des hémoglobinopathies et de l’International Committee for Transfusion Medicine.
Recommandations concernant l’âge du sang :
1. Le jumelage phénotypique doit primer sur l’âge du sang donné lors du choix d’unité pour un patient drépanocytaire.
2. Sur le plan clinique, on ignore l’étendue des avantages et des désavantages liés à l’âge du sang. Par conséquent, il n’est pas recommandé de poser des limites de temps de conservation des unités.
Bibliographie
1. Wahl, Shannon et Keith C. Quirolo. « Current Issues in Blood Transfusion for Sickle Cell Disease », Current Opinion in Pediatrics, vol. 21, no 1, février 2009, p. 15-21.
2. Rees, David C., Thomas N. Williams et Mark T. Gladwin. « Sickle-Cell Disease », The Lancet, vol. 376, no 9757, 11 décembre 2010, p. 2018-2031.
3. Adams, Robert J., Virgil C. McKie, Lewis Hsu, Beatrice Files, Elliott Vichinsky, Charles Pegelow, Miguel Abboud, Dianne Gallagher, Abdullah Kutlar, Fenwick T. Nichols, Duane R. Bonds, Donald Brambilla et coll. « Prevention of a First Stroke by Transfusions in Children with Sickle Cell Anemia and Abnormal Results on Transcranial Doppler Ultrasonography », The New England Journal of Medicine, vol. 339, no 1, 2 juillet 1998, p. 5-11.
4. Adams, Robert J. et Donald Brambilla. « Discontinuing Prophylactic Transfusions Used to Prevent Stroke in Sickle Cell Disease », The New England Journal of Medicine, vol. 353, no 26, 29 décembre 2005, p. 2769-2778.
5. Sarode, Ravindra et Fevzi Altuntas. « Blood Bank Issues Associated with Red Cell Exchanges in Sickle Cell Disease ». Journal of Clinical Apheresis, vol. 21, no 4, 19 décembre 2006, p. 271-273.
6. Swerdlow, Paul S. « Red Cell Exchange in Sickle Cell Disease », Hematology, American Society of Hematology Education Program, 1er janvier 2006, p. 48-53.
7. National Heart, Lung and Blood Institute. Evidence-Based Management of Sickle Cell Disease: Expert Panel Report, [En ligne], 2014. [https://www.nhlbi.nih.gov/health-topics/evidence-based-management-sickle-cell-disease]
8. Wahl, Shannon Kelly, Alicia Garcia, Ward Hagar, Ginny Gildengorin, Keith Quirolo et Elliot Vichinsky. « Lower Alloimmunization Rates in Pediatric Sickle Cell Patients on Chronic Erythrocytapheresis Compared to Chronic Simple Transfusions », Transfusion, vol. 52, no 12, 23 avril 2012, p. 2671-2676.
9. Rosse, Wendell F., Dianne Gallagher, Thomas R. Kinney, Oswaldo Castro, Harvey Dosik, John Moohr, Winfred Wang, Paul S. Levy et la Cooperative Study of Sickle Cell Disease. « Transfusion and Alloimmunization in Sickle Cell Disease », Blood, vol. 76, no 7, 1er octobre 1990, p. 1431-1437.
10. Garratty, George. « Severe Reactions Associated with Transfusion of Patients with Sickle Cell Disease », Transfusion, vol. 37, no 4, avril 1997, p. 357-361.
11. Ness, Paul M. « To Match or not to Match: The Question for Chronically Transfused Patients with Sickle Cell Anemia », Transfusion, vol. 34, no 7, juillet 1994, p. 558-560.
12. Stokes, Ian C., Peter A. Downie, Erica M. Wood, Donald K. Bowden, Paul T. Monagle et Chris D. Barnes. « Hyperhaemolysis in Sickle Cell Disease: An Unusual and Potentially Life-Threatening Complication », The Medical Journal of Australia, vol. 192, no 5, 1er mars 2010, p. 281-282.
13. Aygun Banu, Savitri Padmanabhan, Carole Paley et Visalam Chandrasekaran. « Clinical Significance of RBC Alloantibodies and Autoantibodies in Sickle Cell Patients Who Received Transfusions », Transfusion, vol. 42, no 1, janvier 2002, p. 37-43.
14. Vichinsky, Elliott P., Ann Earles, Robert A. Johnson, M. Silvija Hoag, Amber Williams et Bertram Lubin. « Alloimmunization in Sickle Cell Anemia and Transfusion of Racially Unmatched Blood », The New England Journal of Medicine, vol. 322, no 23, 7 juin 1990, p. 1617-1621.
15. Castro, Oswaldo, S. Gerald Sandler, Patricia Houston-Yu et Sohail Rana. « Predicting the Effect of Transfusing Only Phenotype-Matched RBCs to Patients with Sickle Cell Disease: Theoretical and Practical Implications », Transfusion, vol. 42, no 6, juin 2002, p. 684-690.
16. Tahhan, H. R., C. T. Holbrook, L. R. Braddy, L. D. Brewer et J. D. Christie. « Antigen-Matched Donor Blood in the Transfusion Management of Patients with Sickle Cell Disease », Transfusion, vol. 34, no 7, juillet 1994, p. 562-569.
17. Ameen, Reem, Salem Al ShemmariI et Abdulaziz Al-Bashir. « Red Blood Cell Alloimmunization Among Sickle Cell Kuwaiti Arab Patients Who Received Red Blood Cell Transfusion », Transfusion, vol. 49, no 8, août 2009, p. 1649-1654.
18. Vichinsky, Elliott P., Naomi L. Luban, Elizabeth Wright, Nancy Olivieri, Catherine Driscoll, Charles H. Pegelow et Robert J. Adams. « Prospective RBC Phenotype Matching in a Stroke-Prevention Trial in Sickle Cell Anemia: A Multicenter Transfusion Trial », Transfusion, vol. 41, no 9, septembre 2001, p. 1086-1092.
19. Lee Margaret T., Sergio Piomelli, Suzanne Granger, Scott T. Miller, Shannon Harkness, Donald J. Brambilla, et Robert J. Adams. « Stroke Prevention Trial in Sickle Cell Anemia (STOP): Extended Follow-up and Final Results », Blood, vol. 108, no 3, 1er août 2006, p. 847-852.
20. Lasalle-Williams, Michèle, Rachelle Nuss, Tuan Le, Laura Cole, Kathy Hassell, James R. Murphy et Daniel R. Ambruso. « Extended Red Blood Cell Antigen Matching for Transfusions in Sickle Cell Disease: A Review of a 14-Year Experience from a Single Center (CME) », Transfusion, vol. 51, no 8, août 2011, p. 1732-1739.
21. Fasano, Ross M., Erin K. Meyer, Jane Branscomb, Mia S. White, Robert W. Gibson et James R. Eckman. « Impact of Red Blood Cell Antigen Matching on Alloimmunization and Transfusion Complications in Patients with Sickle Cell Disease: A Systematic Review », Transfusion Medicine Reviews, vol. 33, no 1, janvier 2019, p. 12-23.
22. Karafin, Matthew S., Arun K. Singavi, Mehraboon S. Irani, Kathleen E. Puca, Lisa Baumann Kreuziger, Pippa Simpson et Joshua J. Field. « Red Cell Storage Age Policy for Patients with Sickle Cell Disease: A Survey of Transfusion Service Directors in the United States », Transfusion and Apheresis Science, vol. 54, no 1, février 2016, p. 158-162.
23. Osby, Melanie et Ira A. Shulman. « Phenotype Matching of Donor Red Blood Cell Units for Nonalloimmunized Sickle Cell Disease Patients: A Survey of 1182 North American Laboratories », Archives of Pathology & Laboratory Medicine, vol. 129, no 2, février 2005, p. 190-193.
24. Canadian Haemoglobinopathy Association. [En ligne], Consensus Statement on the Care of Patients with Sickle Cell Disease in Canada, v. 2.0, Ottawa, 2018. [https://www.canhaem.org/wp-content/uploads/2018/05/Sickle-Cell-Consensus.pdf]
25. Compernolle, Veerle, Stella T. Chou, Susano Tanael, William Savage, Jo Howard, Cassandra D. Josephson, Isaac Odame, Christopher Hogan, Gregory Denomme et Nadine Shehata. « Red Blood Cell Specifications for Patients with Hemoglobinopathies: A Systematic Review and Guideline », Transfusion, vol. 58, no 6, juin 2018, p. 1555-1566.
26. Chou, Stella T., Mouaz Alsawas, Ross M. Fasano, Joshua J. Field, Jeanne E. Henrickson, Jo Howard, Michelle Kameka, Janet L. Kwiatkowski, France Pirenne, Patricia A. Shi, Sean R. Stowell, Swee Lay Thein, Connie M. Westhoff, Trisha E. Wong et Elie A. Akl. « American Society of Hematology 2020 Guidelines for Sickle Cell Disease: Transfusion Support », Blood Advances, vol. 4, no 2, 27 janvier 2020, p. 327-355.
27. Tournamille, Christophe, Yves Colin, Jean-Pierre Cartron et Caroline Le Van Kim. « Disruption of a GATA Motif in the Duffy Gene Promoter Abolishes Erythroid Gene Expression in Duffy-Negative Individuals », Nature Genetics, no 10, 1er juin 1995, p. 224-228.
28. Wilkinson, Katie, Samantha Harris, Prashant Gaur, Askale Haile, Rosalind Armour, Gayle Teramura et Meghan Delaney. « Molecular Blood Typing Augments Serologic Testing and Allows for Enhanced Matching of Red Blood Cells for Transfusion in Patients with Sickle Cell Disease », Transfusion, vol. 52, no 2, février 2012, p. 381-388.
29. Lau, W. « Transfusion Support for Sickle Cell Anemia Patients: Memo from Canadian Blood Services Diagnostic Services », Toronto, 2009.
30. Avard, Denise, Linda Kharaboyan et Bartha Maria Knoppers. « Newborn Screening for Sickle Cell Disease: Socio-Ethical Implications », dans MCLEAN, Sheila A. M., éd. First do no harm: Law, ethics and healthcare, Aldershot (R.-U.), Ashgate Publishing Limited, 2006, p. 493-507. [https://papyrus.bib.umontreal.ca/xmlui/bitstream/handle/1866/694/Newborn%20Screening%20for%20SCD?sequence=1]
31. Afeyi-Annan, Araba et Mark E. Brecher. « Pre-Transfusion Phenotype Matching for Sickle Cell Disease Patients », Transfusion, vol. 44, no 4, avril 2004, p. 619-620.
32. Vichinsky, Elliott P. « The Prevention and Management of Alloimmunization in Sickle Cell Disease: The Benefit of Extended Phenotypic Matching of Red Blood Cells », Immunohematology, vol. 28, no 1, 2012, p. 20-23.
33. Winkler, Anne M. et Cassandra D. Josephson. « Transfusion Practices for Patients with Sickle Cell Disease at Major Academic Medical Centers Participating in the Atlanta Sickle Cell Consortium ». Immunohematology, vol. 28, no 1, 2012, p. 24-26.
34. Chou, Stella T. et David F. Friedman. « Transfusion Practices for Patients with Sickle Cell Disease at the Children's Hospital of Philadelphia ». Immunohematology, v. 28, no 1, 2012, p. 27-30.
35. Roberts Dionna O., Brittany Covert, Terianne Lindsey, V. Edwards, Lauren McLaughlin, John Theus, Ricardo J. Wray, Keri Jupka, David Baker, M. Robbins et Michael R. Debaun. « Directed Blood Donor Program Decreases Donor Exposure for Children with Sickle Cell Disease Requiring Chronic Transfusion », Immunohematology, vol. 28, no 1, 2012, p. 7-12.
36. Sloan, Steven R. « Transfusions for Patients with Sickle Cell Disease at Children's Hospital Boston », Immunohematology, vol. 28, no 1, 2012, p. 17-19.
37. Fasano, Ross M., Wendy Paul, Elizabeth Siegal et Naomi L.C. Luban. « Transfusion Protocol for Patients with Sickle Hemoglobinopathies at Children's National Medical Center », Immunohematology, vol. 28, no 1, 2012, p. 13-16.
38. Karafin, Matthew S., R. Sue Shirey, Paul M. Ness, Karen E. King. « Antigen-Matched Red Blood Cell Transfusions for Patients with Sickle Cell Disease at the Johns Hopkins Hospital », Immunohematology, vol. 28, no 1, 2012, p. 3-6.
39. Higgins, John M. et Steven R. Sloan. « Stochastic Modeling of Human RBC Alloimmunization: Evidence for a Distinct Population of Immunologic Responders », Blood, vol. 112, no 6, 2008, p. 2546-2553.
40. Heddle, Nancy M., Richard J. Cook, Donald M. Arnold, Yang Liu, Rebecca Barty, Mark A. Crowther, P.J. Devereaux, Jack Hirsh, Theodore E. Warkentin, Kathryn E. Webert, David Roxby, Magdalena Sobieraj-Teague, Andrea Kurz, Daniel I. Sessler, Priscilla Figueroa, Martin Ellis et John W. Eikelboom. « Effect of Short-Term Vs. Long-Term Blood Storage on Mortality after Transfusion », The New England Journal of Medicine, vol. 375, no 20, 17 novembre 2016, p. 1937-1945.
41. Steiner, Marie E., Paul M. Ness, Susan F. Assmann, Darrell J. Triulzi, Steven R. Sloan, Meghan Delaney, Suzanne Granger, Elliott Bennett-Guerrero, Morris A. Blajchman, Vincent Scavo, Jeffrey L. Carson, Jerrold H. Levy, Glenn Whitman, Pamela D'Andrea, Shelley Pulkrabek, Thomas L. Ortel, Larissa Bornikova, Thomas Raife, Kathleen E. Puca, Richard M. Kaufman, Gregory A. Nuttall, Pampee P. Young, Samuel Yousef, Richard Engelman, Philip E. Greilich, Ronald Miles, Cassandra D. Josephson, Arthur Bracey, Rhonda Cooke, Jeffrey McCullough, Robert Hunsaker, Lynne Uhl, Janice G. McFarland, Yara Park, Melissa M. Cushing, Charles T. Klodell, Ravindra Karanam, Pamela R. Roberts, Cornelius Dyke, Eldad A. Hod et Christopher P. Stowell. « Effects of Red-Cell Storage Duration on Patients Undergoing Cardiac Surgery », The New England Journal of Medicine, vol. 372, no 15, 9 avril 2015, p. 1419-1429.
42. Lacroix, Jacques, Paul C. Hébert, Dean A. Fergusson, Alan Tinmouth, Deborah J. Cook, John C. Marshal, Lucy Clayton, Lauralyn McIntyre, Jeannie Callum, Alexis F. Turgeon, Morris A. Blajchman, Timothy S. Walsh, Simon J. Stanworth, Helen Campbell, Gilles Capellier, Pierre Tiberghien, Laurent Bardiaux, Leo van de Watering, Nardo J. van der Meer, Elham Sabri et Dong Vo. « Age of Transfused Blood in Critically Ill Adults », The New England Journal of Medicine, vol. 372, no 15, 9 avril 2015, p. 1410-1418.
43. Fergusson, Dean A., Paul Hebert, Debora L. Hogan, Louise Lebel, Nicole Rouvinez-Bouali, John A. Smyth, Koravangattu Sankaran, Alan Tinmouth, Morris A. Blajchman, Lajos Kovacs, Christian Lachance, Shoo Lee, C. Robin Walker, Brian Hutton, Robin Ducharme, Katelyn Balchin, Tim Ramsay, Jason C. Ford, Ashok Kakadekar, Kuppuchipalayam Ramesh et Stan Shapiro. « Effect of Fresh Red Blood Cell Transfusions on Clinical Outcomes in Premature, Very Low-Birth-Weight Infants: The ARIPI Randomized Trial », JAMA, vol. 308, no 14, 2012, p. 1443-1451. [https://www.ncbi.nlm.nih.gov/pubmed/23045213]
44. Hess, John R. « Red Cell Changes During Storage », Transfusion and Apheresis Science, vol. 43, no 1, août 2010, p. 51-59.
45. Dhabangi, Aggrey, Brenda Ainomugisha, Christine Cswerti-Gazdewich, Henry Ddungu, Dorothy Kyeyune, Ezra Musisi, Robert Opoka, Christopher P. Stowell et Walter H. Dzik. « Effect of Transfusion of Red Blood Cells with Longer vs Shorter Storage Duration on Elevated Blood Lactate Levels in Children with Severe Anemia: The TOTAL Randomized Clinical Trial », JAMA, vol. 314, no 23, 2015, p. 2514-2523.
46. Fields, Melanie E., Monica L. Hulbert, Ling hen, Ari N. Berlin, Ron Jackups et Philip C. Spinella. « Red Blood Cell Storage Duration Is not Associated with Clinical Outcomes for Acute Chest Syndrome in Children with Sickle Cell Disease », Transfusion, vol. 55, no 11, novembre 2015, p. 2714-2721.
47. Karafin, Matthew S., Erica Carpenter, Amy Pan, Pippa Simpson et Joshua J. Field. « Older Red Cell Units Are Associated with an Increased Incidence of Infection in Chronically Transfused Adults with Sickle Cell Disease », Transfusion and Apheresis Science, vol. 56, no 3, 1er juin 2017, p. 345-351.
48. Desai, Payal C., Allison M. Deal, Emily R. Pfaff, Bahjat Qaquish, Leyna M. Hebden, Yara A. Park et Kenneth I. Ataga. « Alloimmunization Is Associated with Older Age of Transfused Red Blood Cells in Sickle Cell Disease », American Journal of Hematology, vol. 90, no 8, août 2015, p. 691-695.
49. British Committee for Standards in Haematology, Clare Milkins, Jenny Berryman, Carol Cantwell, Chris Elliott, Richard Haggas, Joan Jones, Megan Rowley, Michael Williams et Nay Win. « Guidelines for Pre-Transfusion Compatibility Procedures in Blood Transfusion Laboratories », Transfusion Medicine, vol. 23, no 1, février 2013, p. 3-35. [http://www.ncbi.nlm.nih.gov/pubmed/23216974]